Loading required package: GenomicRanges
Loading required package: stats4
Loading required package: BiocGenerics
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:dplyr':
combine, intersect, setdiff, union
The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabs
The following objects are masked from 'package:base':
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which.max, which.min
Loading required package: S4Vectors
Attaching package: 'S4Vectors'
The following objects are masked from 'package:dplyr':
first, rename
The following object is masked from 'package:tidyr':
expand
The following objects are masked from 'package:base':
expand.grid, I, unname
Loading required package: IRanges
Attaching package: 'IRanges'
The following objects are masked from 'package:dplyr':
collapse, desc, slice
The following object is masked from 'package:purrr':
reduce
Loading required package: GenomeInfoDb
Rows: 214516 Columns: 22
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (7): annot_transcript_id, annot_gene_id, annot_gene_name, gene_novelty,...
dbl (5): n_reads, n_donors, dist_to_CAGE_peak, dist_to_polyA_site, dist_to_...
lgl (10): within_CAGE_peak, within_polyA_site, polyA_motif_found, CAGE_suppo...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
whitelist_support
# A tibble: 214,516 × 22
annot_trans…¹ annot…² annot…³ gene_…⁴ trans…⁵ ISM_s…⁶ n_reads n_don…⁷ dist_…⁸
<chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 ENST00000000… ENSG00… ARF5 Known Known <NA> 825 3 6
2 ENST00000000… ENSG00… M6PR Known Known <NA> 1955 3 19
3 ENST00000000… ENSG00… ESRRA Known Known <NA> 11 3 2
4 ENST00000001… ENSG00… FKBP4 Known Known <NA> 2319 3 -30
5 ENST00000001… ENSG00… CYP26B1 Known Known <NA> 63 3 6497
6 ENST00000002… ENSG00… NDUFAF7 Known Known <NA> 158 3 -19
7 ENST00000002… ENSG00… FUCA2 Known Known <NA> 663 3 -10
8 ENST00000002… ENSG00… DBNDD1 Known Known <NA> 421 3 0
9 ENST00000002… ENSG00… HS3ST1 Known Known <NA> 1351 3 -8
10 ENST00000003… ENSG00… CYP51A1 Known Known <NA> 6691 3 -22
# … with 214,506 more rows, 13 more variables: within_CAGE_peak <lgl>,
# dist_to_polyA_site <dbl>, within_polyA_site <lgl>, polyA_motif <chr>,
# dist_to_polyA_motif <dbl>, polyA_motif_found <lgl>, CAGE_support_100 <lgl>,
# CAGE_support_250 <lgl>, CAGE_support_500 <lgl>, PAS_motif_support_35 <lgl>,
# PAS_motif_support_50 <lgl>, PAS_motif_support_100 <lgl>,
# long_read_db <lgl>, and abbreviated variable names ¹annot_transcript_id,
# ²annot_gene_id, ³annot_gene_name, ⁴gene_novelty, ⁵transcript_novelty, …
sanity_check = whitelist_support %>%left_join(sqanti, by =c("annot_transcript_id"="isoform"))sanity_check %>%filter(within_CAGE_peak.x != within_CAGE_peak.y)
# A tibble: 3,163 × 69
annot_trans…¹ annot…² annot…³ gene_…⁴ trans…⁵ ISM_s…⁶ n_reads n_don…⁷ dist_…⁸
<chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 ENST00000070… ENSG00… PKP2 Known Known <NA> NA NA NA
2 ENST00000218… ENSG00… STAG2 Known Known <NA> NA NA NA
3 ENST00000238… ENSG00… ZC2HC1C Known Known <NA> NA NA NA
4 ENST00000245… ENSG00… RPL23 Known Known <NA> NA NA NA
5 ENST00000252… ENSG00… FUT5 Known Known <NA> NA NA NA
6 ENST00000254… ENSG00… UBE2L2 Known Known <NA> NA NA NA
7 ENST00000254… ENSG00… ZSWIM4 Known Known <NA> NA NA NA
8 ENST00000254… ENSG00… AL3919… Known Known <NA> NA NA NA
9 ENST00000261… ENSG00… FOXN3 Known Known <NA> NA NA NA
10 ENST00000262… ENSG00… IKZF4 Known Known <NA> NA NA NA
# … with 3,153 more rows, 60 more variables: within_CAGE_peak.x <lgl>,
# dist_to_polyA_site.x <dbl>, within_polyA_site.x <lgl>, polyA_motif.x <chr>,
# dist_to_polyA_motif <dbl>, polyA_motif_found.x <lgl>,
# CAGE_support_100 <lgl>, CAGE_support_250 <lgl>, CAGE_support_500 <lgl>,
# PAS_motif_support_35 <lgl>, PAS_motif_support_50 <lgl>,
# PAS_motif_support_100 <lgl>, long_read_db <lgl>, chrom <chr>, strand <chr>,
# length <dbl>, exons <dbl>, structural_category <chr>, …
Rows: 214516 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (2): transcript_ID, CAGE_support
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 214516 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (2): transcript_ID, CAGE_support
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 214516 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (2): transcript_ID, CAGE_support
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 214516 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (2): transcript_ID, CAGE_support
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 214516 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (2): transcript_ID, CAGE_support
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 214516 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (2): transcript_ID, CAGE_support
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
support_for_plot
# A tibble: 214,516 × 10
transcript_id trans…¹ withi…² withi…³ withi…⁴ withi…⁵ withi…⁶ withi…⁷ withi…⁸
<chr> <fct> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl>
1 ENST00000494… Known FALSE FALSE FALSE FALSE FALSE FALSE FALSE
2 ENST00000424… Known FALSE FALSE FALSE FALSE FALSE FALSE FALSE
3 ENST00000445… Known FALSE FALSE FALSE FALSE FALSE FALSE FALSE
4 ENST00000457… Known TRUE TRUE TRUE TRUE FALSE FALSE TRUE
5 ENST00000414… Known TRUE FALSE FALSE FALSE FALSE FALSE TRUE
6 ENST00000655… Known FALSE TRUE TRUE TRUE FALSE FALSE FALSE
7 ENST00000457… Known FALSE TRUE TRUE TRUE FALSE FALSE FALSE
8 ENST00000591… Known FALSE FALSE FALSE FALSE FALSE FALSE TRUE
9 ENST00000644… Known FALSE FALSE FALSE FALSE FALSE FALSE FALSE
10 ENST00000445… Known TRUE TRUE TRUE TRUE FALSE FALSE TRUE
# … with 214,506 more rows, 1 more variable: polyA_motif_found <lgl>, and
# abbreviated variable names ¹transcript_novelty, ²within_CAGE_refTSS,
# ³within_CAGE_fetal, ⁴within_ATAC_Greenleaf, ⁵within_ATAC_Nowakowski,
# ⁶within_ATAC_LuisCP, ⁷within_ATAC_LuisGZ, ⁸within_polyA_site
# A tibble: 28 × 6
transcript_novelty n name value prop end
<fct> <int> <fct> <int> <dbl> <fct>
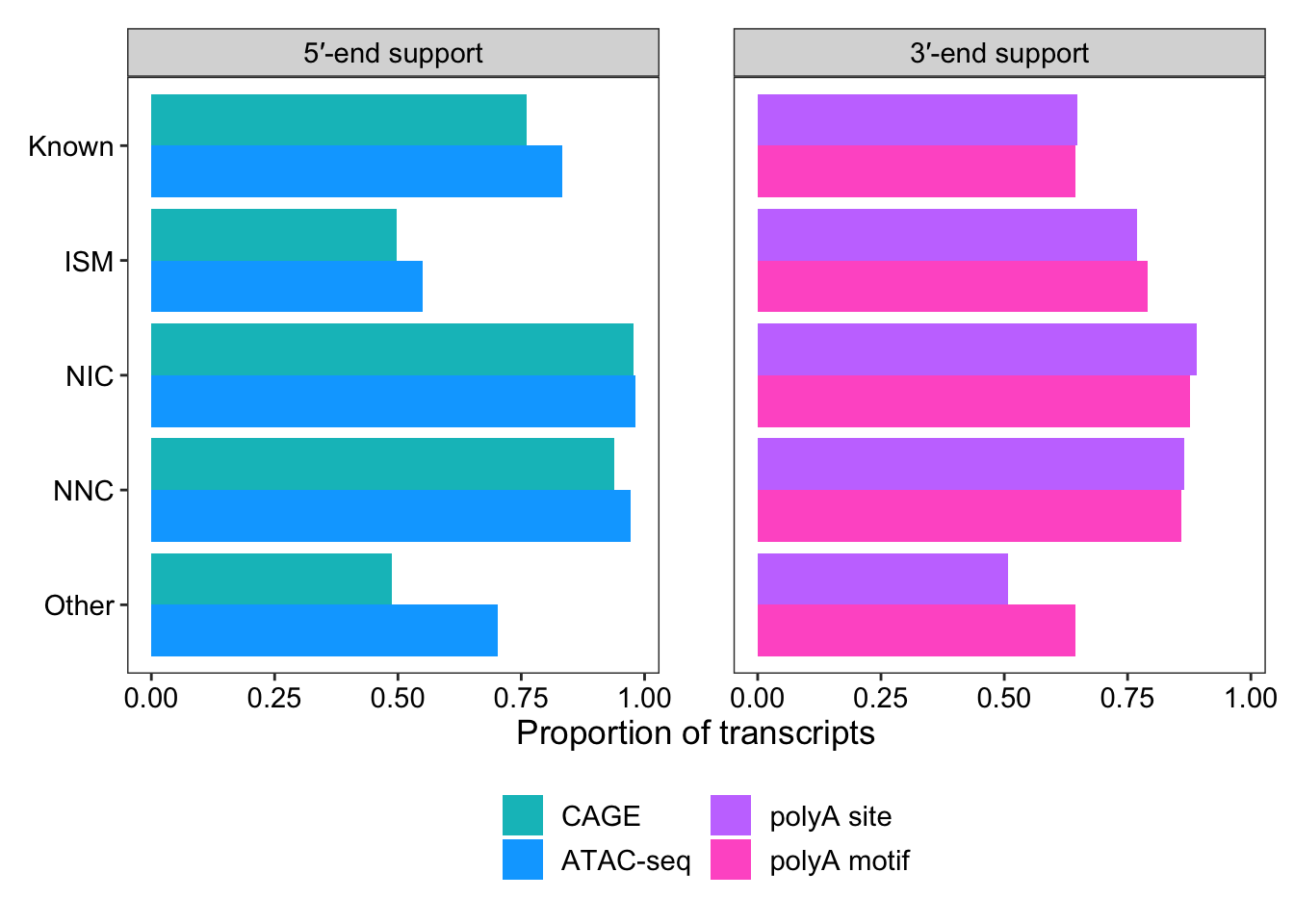
1 ISM 83089 within_CAGE_peak 41387 0.498 5′-end support
2 ISM 83089 within_ATAC_peak 45668 0.550 5′-end support
3 ISM 83089 within_polyA_site 63833 0.768 3′-end support
4 ISM 83089 polyA_motif_found 65760 0.791 3′-end support
5 Known 65006 within_CAGE_peak 49495 0.761 5′-end support
6 Known 65006 within_ATAC_peak 54138 0.833 5′-end support
7 Known 65006 within_polyA_site 42187 0.649 3′-end support
8 Known 65006 polyA_motif_found 41856 0.644 3′-end support
9 NIC 50621 within_CAGE_peak 49442 0.977 5′-end support
10 NIC 50621 within_ATAC_peak 49674 0.981 5′-end support
# … with 18 more rows
# A tibble: 4 × 3
transcript_novelty `within_polyA_site | polyA_motif_found` n
<fct> <lgl> <int>
1 ISM FALSE 9997
2 ISM TRUE 73092
3 Known FALSE 15470
4 Known TRUE 49536
Rows: 474236 Columns: 9
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (1): chrom
dbl (5): genomic_start_coord, genomic_end_coord, strand, X5, X6
lgl (3): X7, X8, X9
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 383616 Columns: 9
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (1): chrom
dbl (5): genomic_start_coord, genomic_end_coord, strand, X5, X6
lgl (3): X7, X8, X9
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 4974342 Columns: 10
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (1): chrom
dbl (8): genomic_start_coord, genomic_end_coord, strand, X5, X6, X7, X8, X9
lgl (1): X10
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.