Rows: 10809 Columns: 11
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (2): gene_name, gene_id
dbl (6): DTU_qval_min, DTU_pval_min, DTE_qval_min, DTE_pval_min, DGE_pval, D...
lgl (3): DTU, DTE, DGE
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
DTU <- tableS3.gene %>%filter(DTU==T) %>%arrange(DTU_qval_min) %>% dplyr::select(gene_id) %>%pull() %>%substr(0,15)DTU.bg = tableS3.isoform%>% dplyr::select(gene_id)%>%unique() %>%pull() %>%substr(0,15)## Pathway analysis with gProfileR## Note: ordered query here because genes are ranked by DTU significance. Usually this will be F## Always use the matching background gene setpath = gprofiler2::gost(query=DTU,ordered_query = T,correction_method ='fdr',custom_bg = DTU.bg, sources =c("GO","KEGG", "REACTOME"))
Detected custom background input, domain scope is set to 'custom'
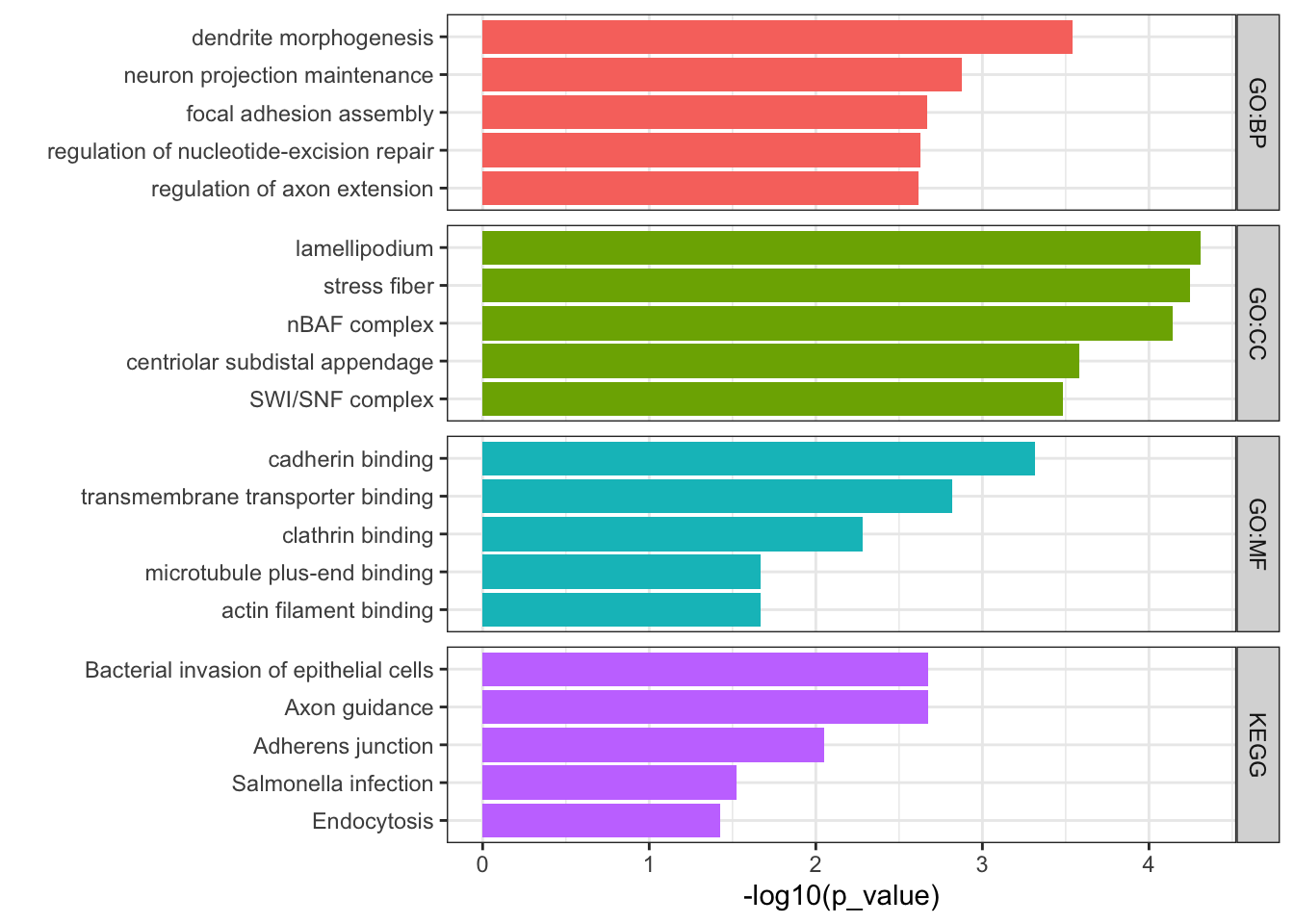
## Filter results for terms between 5-3000 genes. Also here I remove results where a child GO term is also included, just because there were a lot of results. Can remove this df_path =as_tibble(path$result) %>%filter(term_size <3000, path$result$term_size>5) df_path <- df_path %>%filter(!term_id %in%unlist(df_path$parents))## Plot top 5 results per databaseFig3d <- df_path %>%group_by(source) %>%slice_min(p_value,n=5,with_ties = T) %>%ungroup() %>%ggplot(aes(x=reorder(term_name, -p_value), y=-log10(p_value), fill=source)) +geom_bar(stat='identity',position=position_identity()) +coord_flip() +theme_bw() +labs(x="") +facet_grid(source~., space ='free', scales='free') +theme(legend.position ='none')Fig3d
DTU.up <- tableS3.isoform %>%filter(DTU_dIF >0, DTU_qval < .05) %>%arrange(DTU_qval) %>% dplyr::select(gene_id)%>%unique() %>%pull() %>%substr(0,15) DTU.down <- tableS3.isoform %>%filter(DTU_dIF <0, DTU_qval < .05) %>%arrange(DTU_qval) %>% dplyr::select(gene_id)%>%unique() %>%pull() %>%substr(0,15)DTU.bg = tableS3.isoform %>%arrange(DTU_qval) %>% dplyr::select(gene_id)%>%unique() %>%pull() %>%substr(0,15)## Pathway analysis with gProfileR## Note: ordered query here because genes are ranked by DTU significance. Usually this will be F## Always use the matching background gene setpath.up = gprofiler2::gost(query=DTU.up,ordered_query = T,correction_method ='fdr',custom_bg = DTU.bg, sources =c("GO","KEGG", "REACTOME", "CORUM", "WP"))
Detected custom background input, domain scope is set to 'custom'
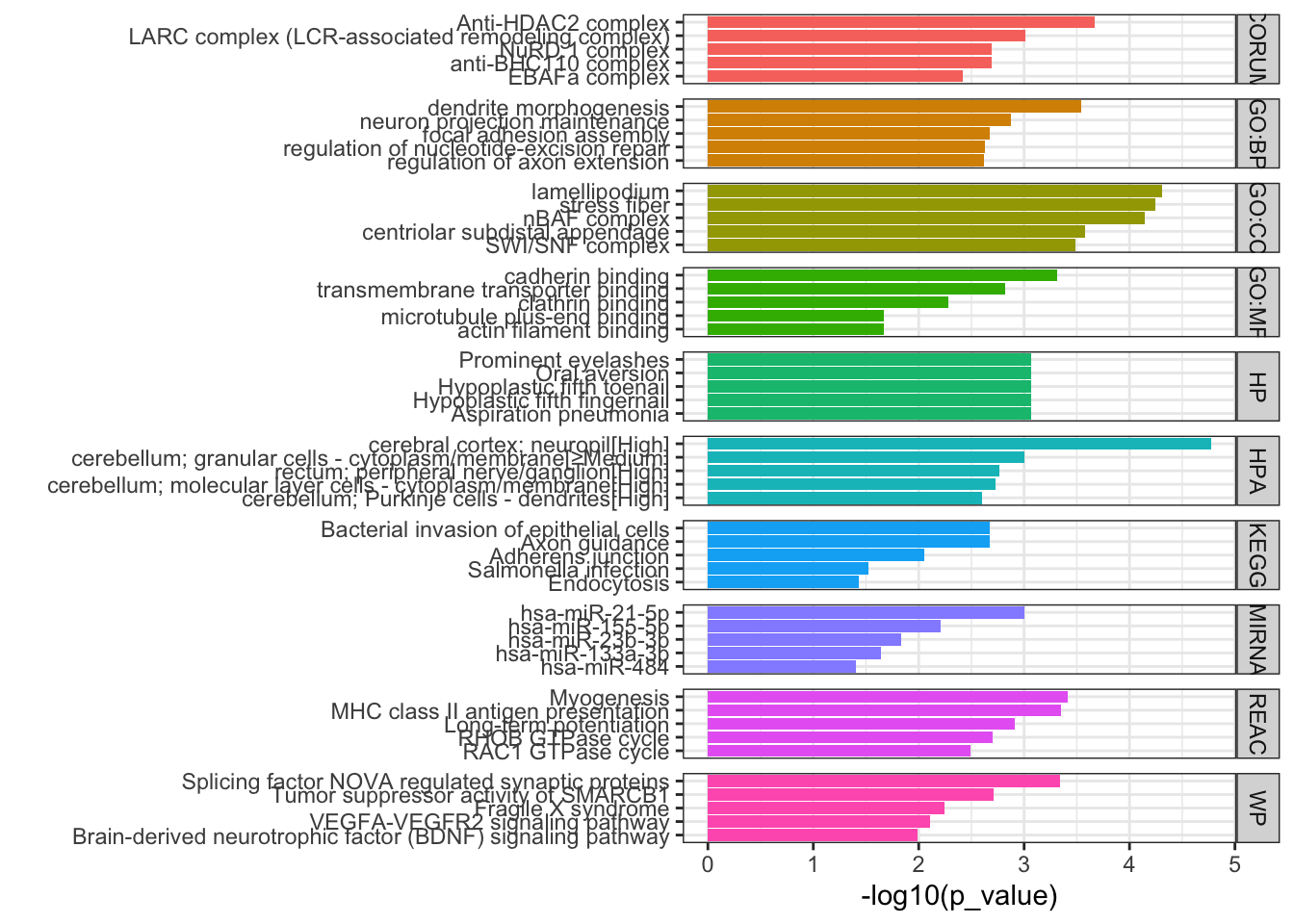
## Filter results for terms between 5-3000 genes. Also here I remove results where a child GO term is also included, just because there were a lot of results. Can remove this df_path.up =as_tibble(path.up$result) %>%filter(term_size <3000, term_size>5) %>%mutate(direction='up')df_path.up <- df_path.up %>%filter(!term_id %in%unlist(df_path.up$parents))## Plot top 5 results per databaseFig3d.up <- df_path.up %>%group_by(source) %>%slice_min(p_value,n=8) %>%ungroup() %>%ggplot(aes(x=reorder(term_name, -p_value), y=-log10(p_value), fill=source)) +geom_bar(stat='identity', position=position_identity()) +coord_flip() +theme_bw() +labs(x="") +ggtitle("DTU.up") +facet_grid(source~., space ='free', scales='free') +theme(legend.position ='none')path.down = gprofiler2::gost(query=DTU.down,ordered_query = T,correction_method ='fdr',custom_bg = DTU.bg, sources =c("GO","KEGG", "REACTOME"))
Detected custom background input, domain scope is set to 'custom'
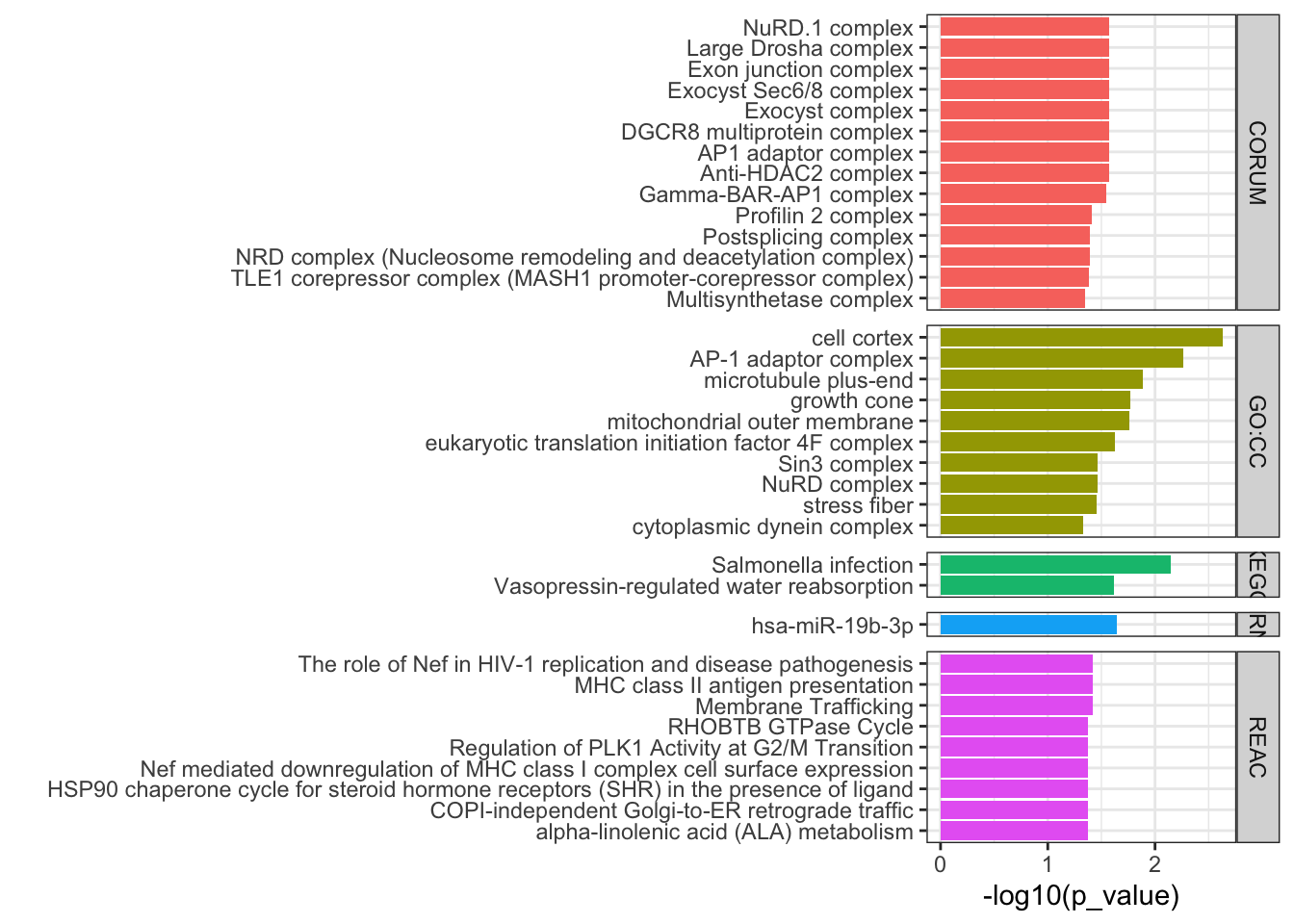
## Filter results for terms between 5-3000 genes. Also here I remove results where a child GO term is also included, just because there were a lot of results. Can remove this df_path.down =as_tibble(path.down$result) %>%filter(term_size <3000, term_size>5) %>%mutate(direction='down')df_path.down <- df_path.down %>%filter(!term_id %in%unlist(df_path$parents))## Plot top 5 results per databaseFig3d.down <- df_path.down %>%group_by(source) %>%slice_min(p_value,n=8) %>%ungroup() %>%ggplot(aes(x=reorder(term_name, -p_value), y=-log10(p_value), fill=source)) +geom_bar(stat='identity', position=position_identity()) +coord_flip() +theme_bw() +labs(x="") +ggtitle("DTU.down") +facet_grid(source~., space ='free', scales='free') +theme(legend.position ='none')cowplot::plot_grid(Fig3d.down, Fig3d.up,ncol=2)
Warning: One or more parsing issues, see `problems()` for details
Rows: 18321 Columns: 26
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (8): gene_id, group, OR (PTV), OR (Class I), OR (Class II), OR (PTV) up...
dbl (16): Case PTV, Ctrl PTV, Case mis3, Ctrl mis3, Case mis2, Ctrl mis2, P ...
lgl (2): De novo mis3, De novo mis2
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 119958 Columns: 20
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (4): gene_id, group, damaging_missense_fisher_gnom_non_psych_OR, ptv_fi...
dbl (16): n_cases, n_controls, damaging_missense_case_count, damaging_missen...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 71456 Columns: 12
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (2): gene_id, group
dbl (9): xcase_lof, xctrl_lof, pval_lof, xcase_mpc, xctrl_mpc, pval_mpc, xca...
lgl (1): pval_infrIndel
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.