suppressPackageStartupMessages({
library(tidyverse)
library(ggrepel)
library(rtracklayer)
})Figure S9 - G, H
base_size_pt = 15
theme_set(theme_gray(base_size = base_size_pt))
theme_update(
plot.title = element_text(size = rel(1.4), hjust = 0.5),
axis.title = element_text(size = rel(1.2)),
axis.text = element_text(color="black", size = rel(1)),
legend.title = element_text(size = rel(1.2)),
legend.text = element_text(color="black", size = rel(1))
)
base_size_mm = base_size_pt * 25.4 / 72.27colors = c(
"Known" = "#009E73",
"ISM" = "#0072B2",
"ISM_Prefix" = "#0072B2",
"ISM_Suffix" = "#0072B2",
"ISM_Both" = "#0072B2",
"NIC" = "#D55E00",
"NNC" = "#E69F00",
"Other" = "#000000"
)source("code/talon_novelty_to_factor.R")support_data = read_tsv(
"data/Fig_S9H/Isoform_counts_4281_knownCells.tsv.gz",
col_select = c(annot_transcript_id, transcript_novelty, ISM_subtype)
)Rows: 137604 Columns: 3
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (3): annot_transcript_id, transcript_novelty, ISM_subtype
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.TSS/TES from external
TSS_support_external = read_tsv(
"data/Fig_S9H/sciso_TSS_support_from_external.w_bulk.txt.gz",
col_types = "ciic_cc",
col_names = c("seqname", "start", "end", "transcript_id", "strand", "overlap")
)TES_support_external = read_tsv(
"data/Fig_S9H/sciso_TES_support_from_external.w_bulk.txt.gz",
col_types = "ciic_cc",
col_names = c("seqname", "start", "end", "transcript_id", "strand", "overlap")
)support_data %>% count()# A tibble: 1 × 1
n
<int>
1 137604TSS_support_external %>% distinct() %>% count(overlap)# A tibble: 15 × 2
overlap n
<chr> <int>
1 AdultCTX.collapsed_classification.filtered_lite_hg19.first_exons.pad_… 76685
2 chess3.0.1_hg19.first_exons.pad_100.bed 97850
3 compare.annotated.first_exons.pad_100.bed 85618
4 compare.combined.first_exons.pad_100.bed 131277
5 cp_vz_0.75_min_7_recovery_talon.first_exons.pad_100.bed 125829
6 FetalCTX.collapsed_classification.filtered_lite_hg19.first_exons.pad_… 78751
7 FetalHIP.collapsed_classification.filtered_lite_hg19.first_exons.pad_… 50933
8 FetalSTR.collapsed_classification.filtered_lite_hg19.first_exons.pad_… 53174
9 gencode.v43lift37.annotation.first_exons.pad_100.bed 114472
10 GSE192955_30ClontechTissue_1D_cDNA_N2_R0_updated.first_exons.pad_100.… 115643
11 GSE192955_PC3E_GS689_HEK293T_1D_cDNA_N2_R0_updated.first_exons.pad_10… 110068
12 GSE192955_PC3E_GS689_LRCA_N2_R0_updated.first_exons.pad_100.bed 89929
13 GTX_flair_filter_transcripts_hg19.first_exons.pad_100.bed 85618
14 HumanCTX.collapsed_classification.filtered_lite_hg19.first_exons.pad_… 92727
15 NA12878-DirectRNA-minimap2-2.5_hg19.first_exons.pad_100.bed 105369TES_support_external %>% distinct() %>% count(overlap)# A tibble: 15 × 2
overlap n
<chr> <int>
1 AdultCTX.collapsed_classification.filtered_lite_hg19.last_exons.pad_2… 54937
2 chess3.0.1_hg19.last_exons.pad_200.bed 70899
3 compare.annotated.last_exons.pad_200.bed 58988
4 compare.combined.last_exons.pad_200.bed 115748
5 cp_vz_0.75_min_7_recovery_talon.last_exons.pad_200.bed 103513
6 FetalCTX.collapsed_classification.filtered_lite_hg19.last_exons.pad_2… 55183
7 FetalHIP.collapsed_classification.filtered_lite_hg19.last_exons.pad_2… 38029
8 FetalSTR.collapsed_classification.filtered_lite_hg19.last_exons.pad_2… 39645
9 gencode.v43lift37.annotation.last_exons.pad_200.bed 95161
10 GSE192955_30ClontechTissue_1D_cDNA_N2_R0_updated.last_exons.pad_200.b… 101137
11 GSE192955_PC3E_GS689_HEK293T_1D_cDNA_N2_R0_updated.last_exons.pad_200… 93976
12 GSE192955_PC3E_GS689_LRCA_N2_R0_updated.last_exons.pad_200.bed 73339
13 GTX_flair_filter_transcripts_hg19.last_exons.pad_200.bed 58988
14 HumanCTX.collapsed_classification.filtered_lite_hg19.last_exons.pad_2… 64272
15 NA12878-DirectRNA-minimap2-2.5_hg19.last_exons.pad_200.bed 78035support_data2 = support_data %>%
left_join(
TSS_support_external %>%
distinct() %>% # dups missed by command-line uniq?
mutate(val = T) %>%
pivot_wider(names_from = overlap, values_from = val) %>%
mutate(across(ends_with(".bed"), ~!is.na(.x))) %>%
select(transcript_id, ends_with(".bed")),
by = c("annot_transcript_id" = "transcript_id")
) %>%
left_join(
TES_support_external %>%
distinct() %>% # dups missed by command-line uniq?
mutate(val = T) %>%
pivot_wider(names_from = overlap, values_from = val) %>%
mutate(across(ends_with(".bed"), ~!is.na(.x))) %>%
select(transcript_id, ends_with(".bed")),
by = c("annot_transcript_id" = "transcript_id")
) %>%
mutate(across(ends_with(".bed"), ~!is.na(.x)))write_tsv(support_data2, "output/figures/revision1/scIso_TSS_TES_external_support.w_bulk.tsv")# Compute percentages
freqs = bind_rows(
support_data2 %>%
talon_novelty_to_factor(split_ISMs = T) %>%
mutate(support = if_any(contains("first_exons"))) %>%
dplyr::count(support, transcript_novelty) %>%
group_by(transcript_novelty) %>%
mutate(freq = n / sum(n), total = sum(n)) %>%
mutate(percent = round(freq*100)) %>%
mutate(percent = if_else(support, percent, NA_real_)) %>%
mutate(tcolor_grp = factor(if_else(percent > 20, "white", "black"))) %>%
mutate(support_type = "5' support in external+bulk"),
support_data2 %>%
talon_novelty_to_factor(split_ISMs = T) %>%
mutate(support = if_any(contains("last_exons"))) %>%
dplyr::count(support, transcript_novelty) %>%
group_by(transcript_novelty) %>%
mutate(freq = n / sum(n), total = sum(n)) %>%
mutate(percent = round(freq*100)) %>%
mutate(percent = if_else(support, percent, NA_real_)) %>%
mutate(tcolor_grp = factor(if_else(percent > 20, "white", "black"))) %>%
mutate(support_type = "3' support in external+bulk")
) %>% mutate(support_type = as_factor(support_type))
xlabel = "Isoform category"
ylabel = "Number of isoforms"
title = "Isoforms in scIso-Seq"
ylabels = waiver()
ymax = 50000
label_pad = max(freqs$total)*.07
ggplot(freqs, aes(x = transcript_novelty %>% fct_rev(), y = n, fill = transcript_novelty,
alpha = support)) +
geom_bar(stat="identity", color = "black") +
xlab(xlabel) + ylab(ylabel) + ggtitle(title) +
scale_fill_manual("", values = colors) +
scale_alpha_manual(values=c(0,1), name = "CAGE support") +
coord_flip(ylim=c(0,ymax)) + guides(fill="none", alpha = "none") +
geom_text(aes(y = total + label_pad,
label = paste0(percent, "%"), color = transcript_novelty),
position = position_dodge(0), size = base_size_mm) +
scale_color_manual(values = colors) +
guides(colour="none", fill="none") +
scale_x_discrete(labels = c("ISM_Prefix" = "ISM Prefix", "ISM_Suffix" = "ISM Suffix")) +
scale_y_continuous(labels = ylabels, expand = c(0, 0)) +
facet_grid(rows = vars(support_type)) +
theme_bw(base_size = base_size_pt) +
theme(axis.line.x = element_line(color="black", size = 0.5),
axis.line.y = element_line(color="black", size = 0.5),
axis.text.x = element_text(color="black", size = base_size_pt),
axis.text.y = element_text(color="black", size = base_size_pt),
axis.title.x = element_text(color="black", size = base_size_pt*1.2),
axis.title.y = element_text(color="black", size = base_size_pt*1.2),
strip.text = element_text(color="black", size = base_size_pt)) +
theme(panel.grid.major = element_blank(),
panel.grid.minor = element_blank())Warning: The `size` argument of `element_line()` is deprecated as of ggplot2 3.4.0.
ℹ Please use the `linewidth` argument instead.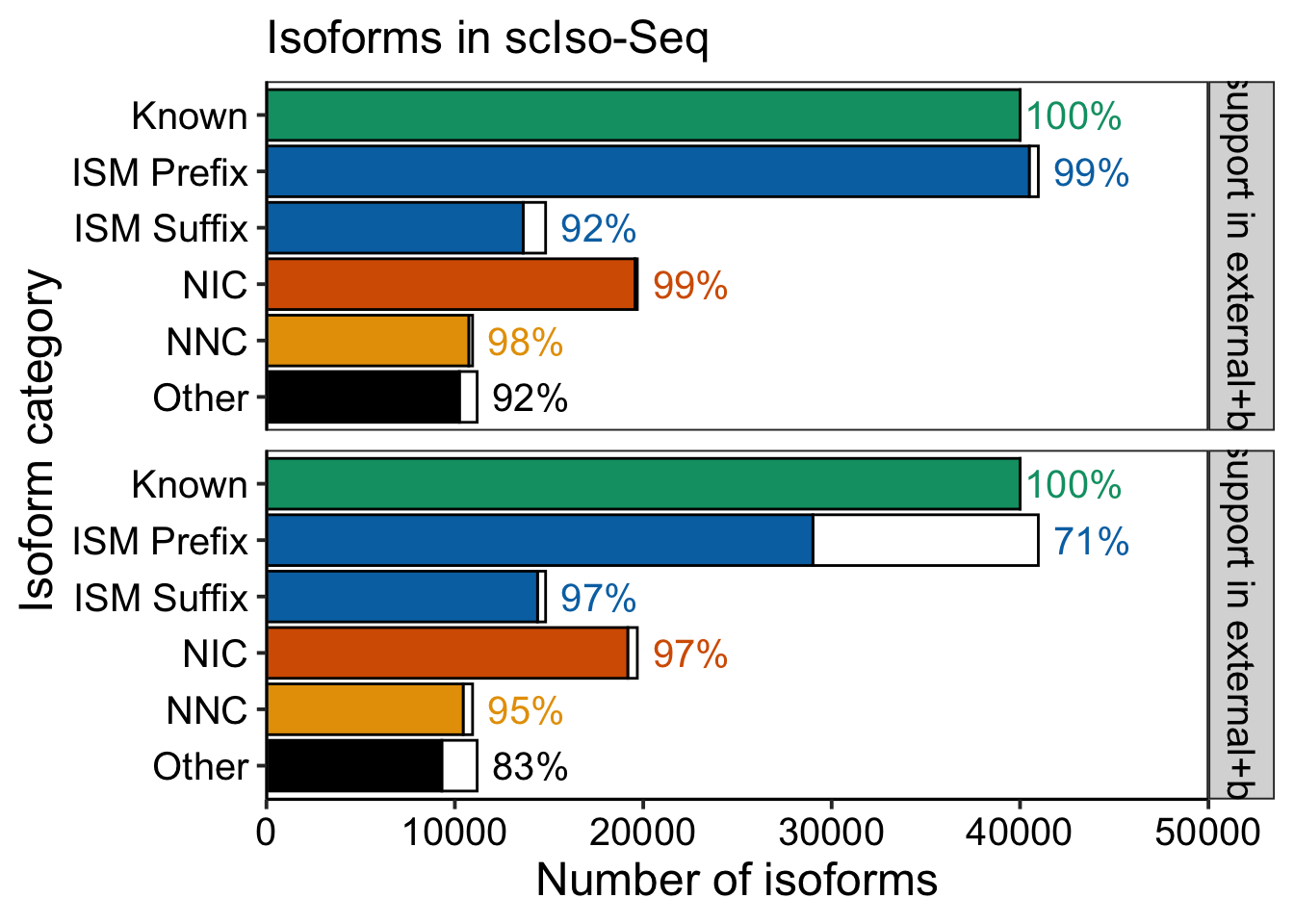
ggsave("output/figures/revision1/FigS9H_for_revision_w_bulk_split.pdf", width = 7.5, height = 6.5)Remake S9G split
iso_types = support_data %>%
talon_novelty_to_factor(split_ISMs = T) %>%
count(transcript_novelty) %>%
mutate(prop = n / sum(n))
ggplot(iso_types, aes(x = transcript_novelty, y = n, fill = transcript_novelty)) +
geom_col() +
geom_text(
aes(label = scales::percent(prop, accuracy = 1)),
vjust = 1.5,
colour = "white",
size = base_size_mm
) +
scale_fill_manual(
values = colors
) +
scale_x_discrete(
labels = c("ISM_Prefix" = "ISM\nPrefix", "ISM_Suffix" = "ISM\nSuffix")
) +
guides(fill = "none") +
xlab("Classification") +
#ylab(expression(paste("Number of transcripts (x", 10^3, ")")))
ylab("Number of isoforms")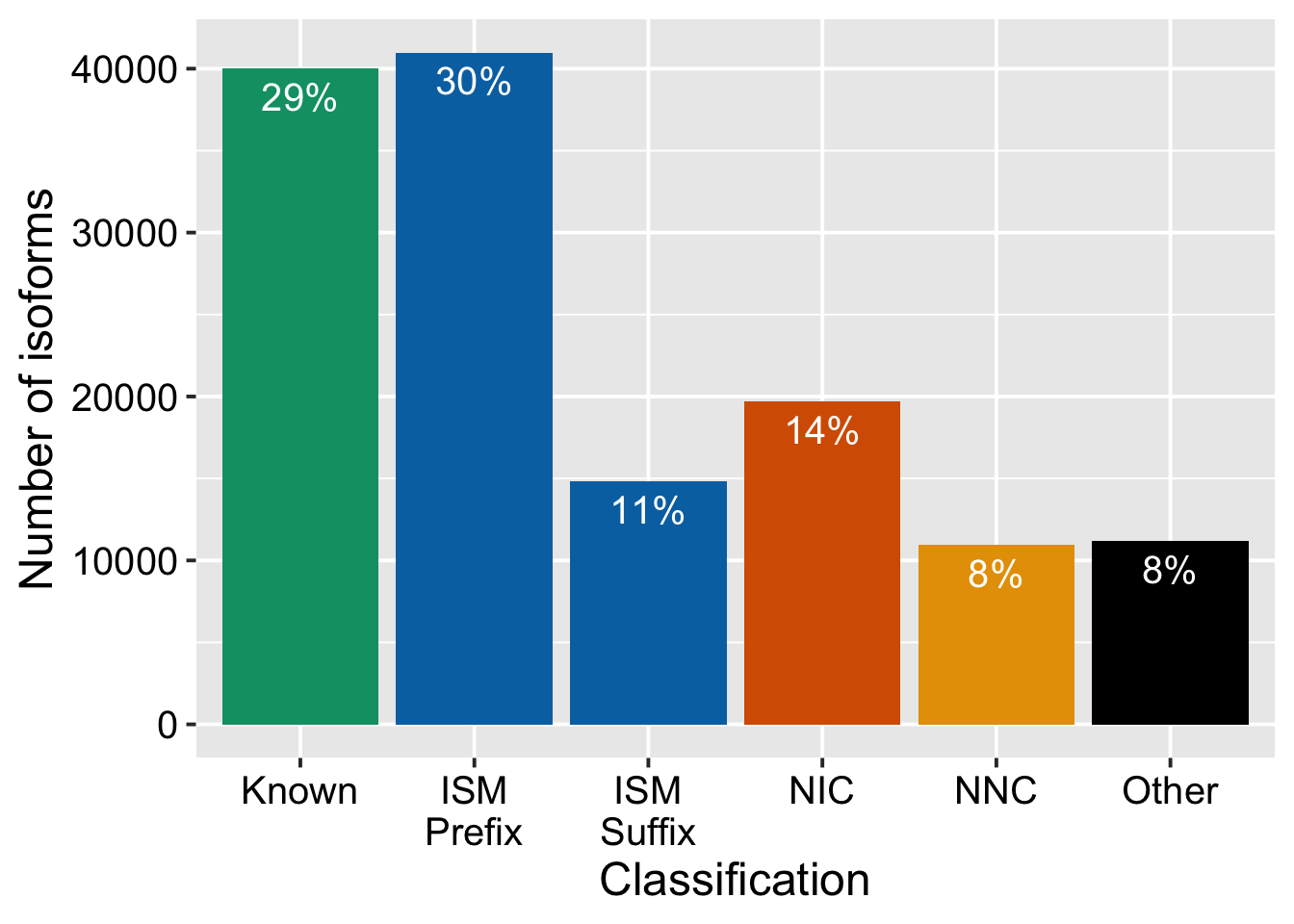
ggsave("output/figures/revision1/FigS9G_for_revision_split.pdf", width = 6, height = 5)