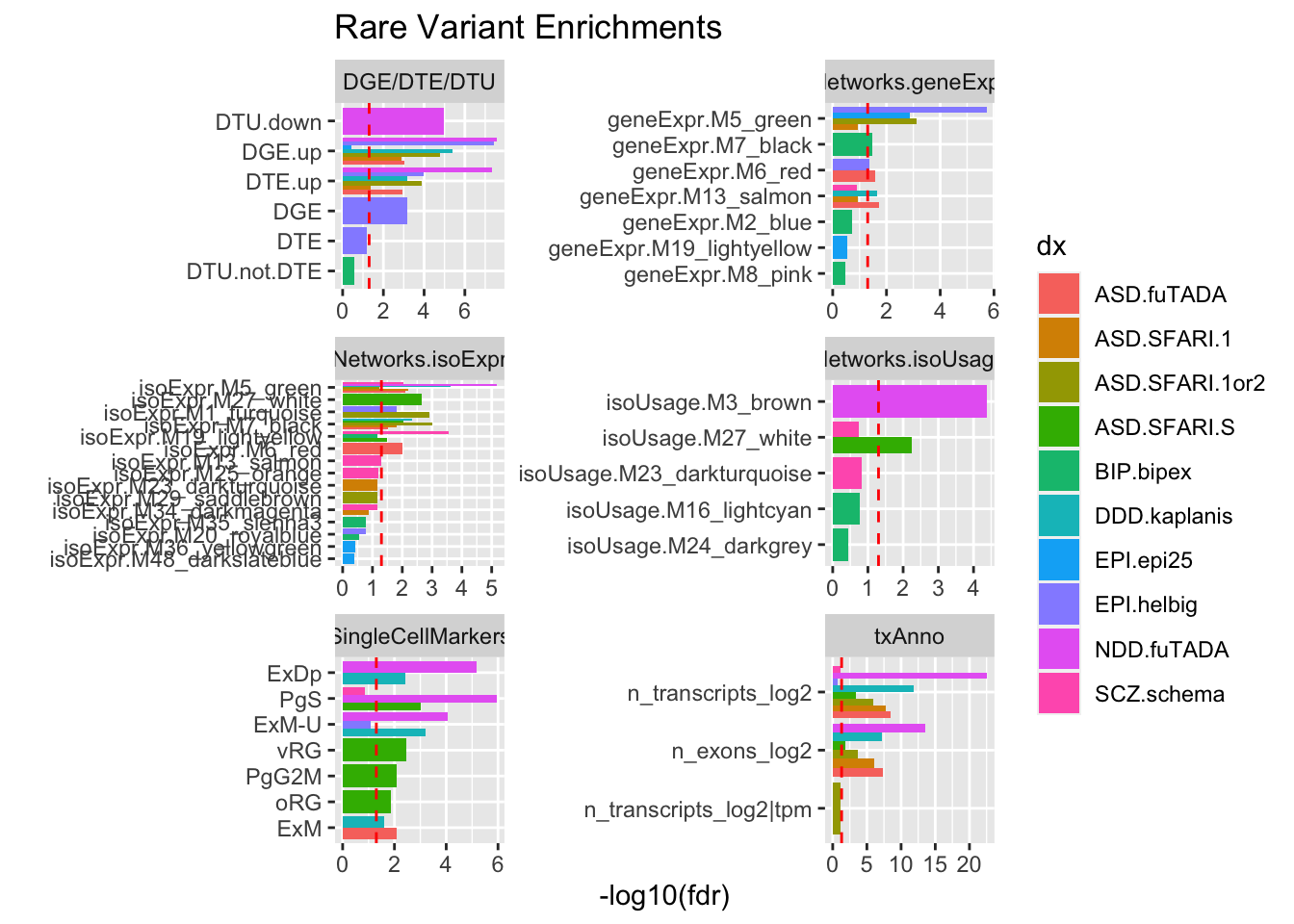
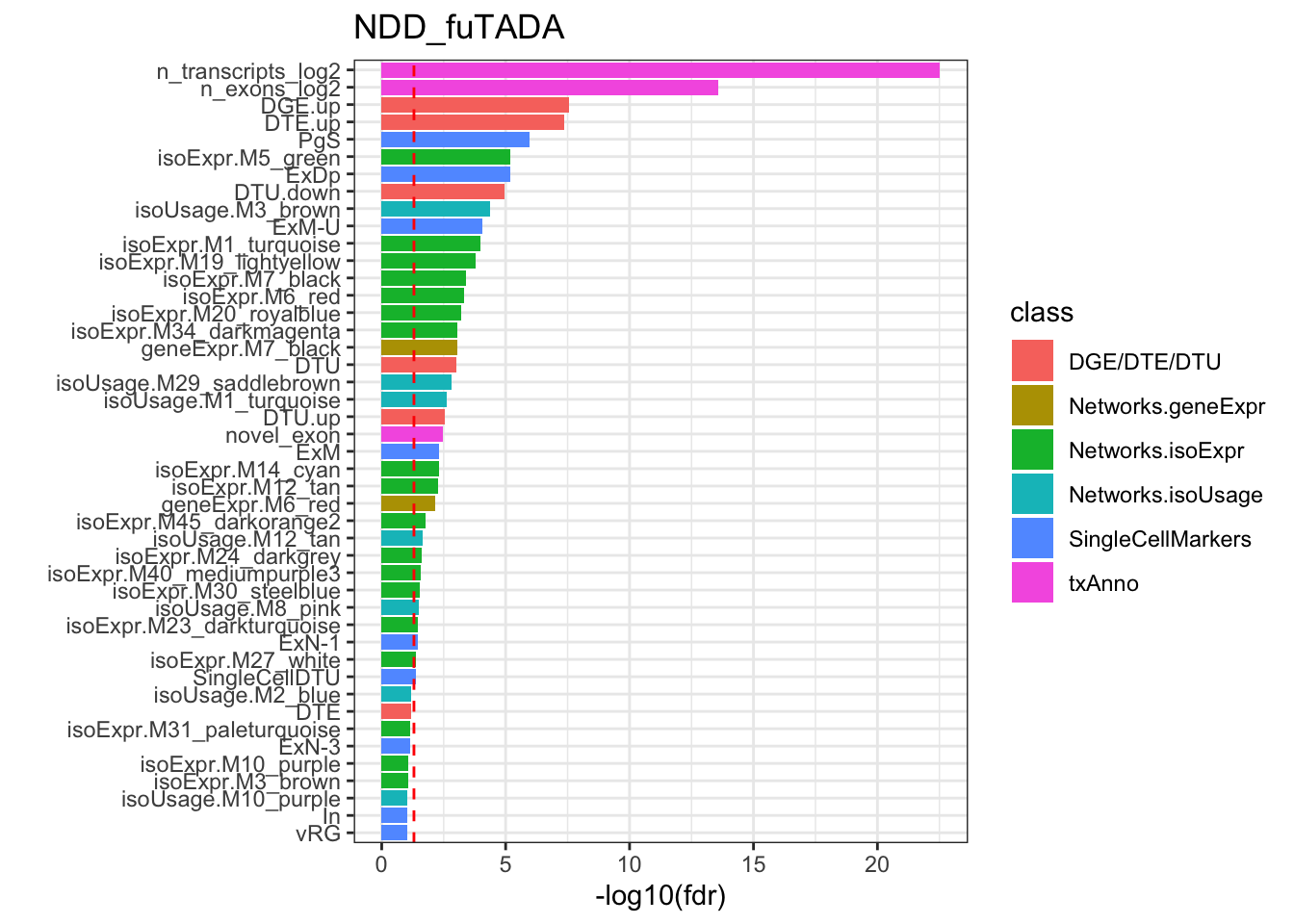
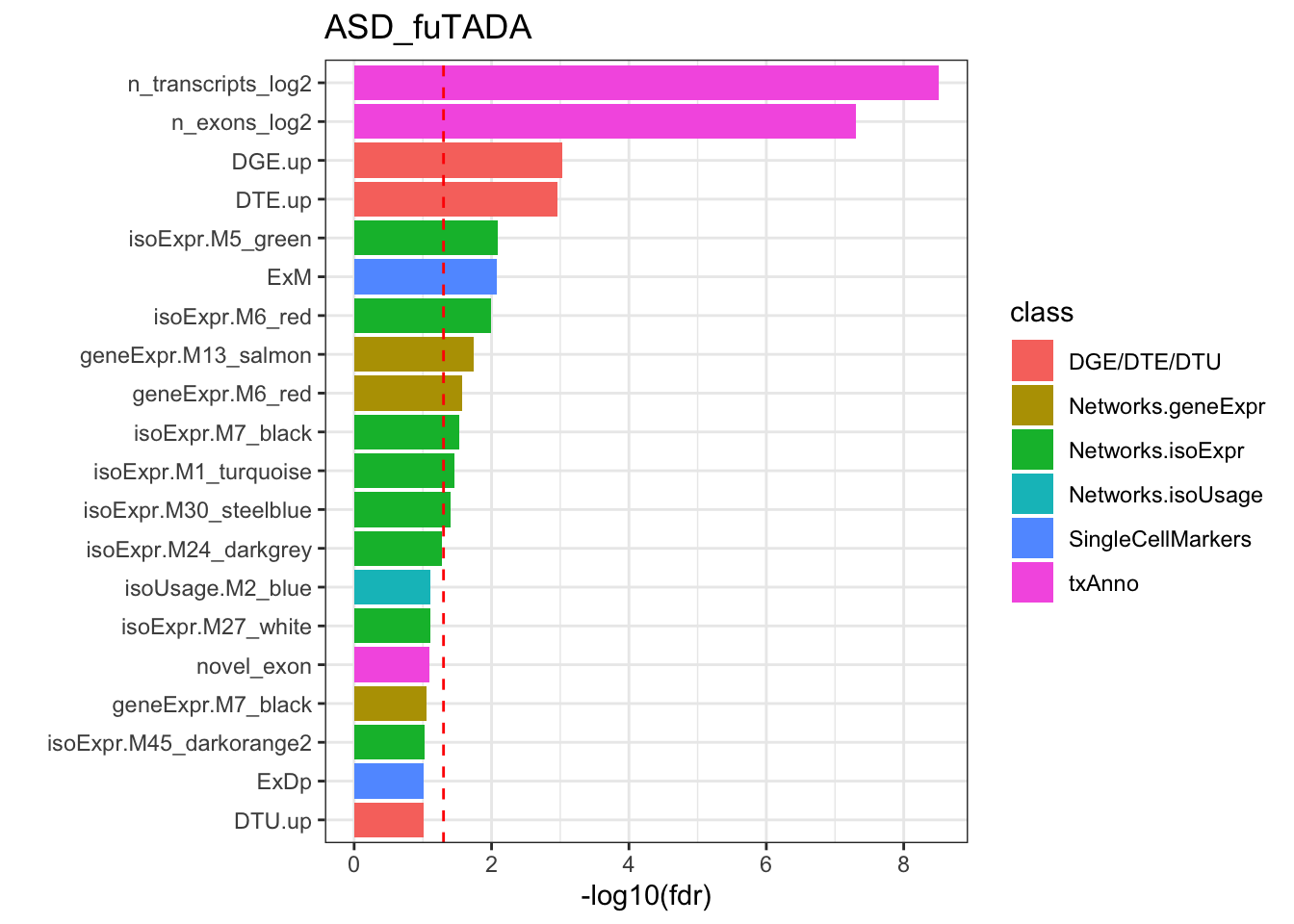
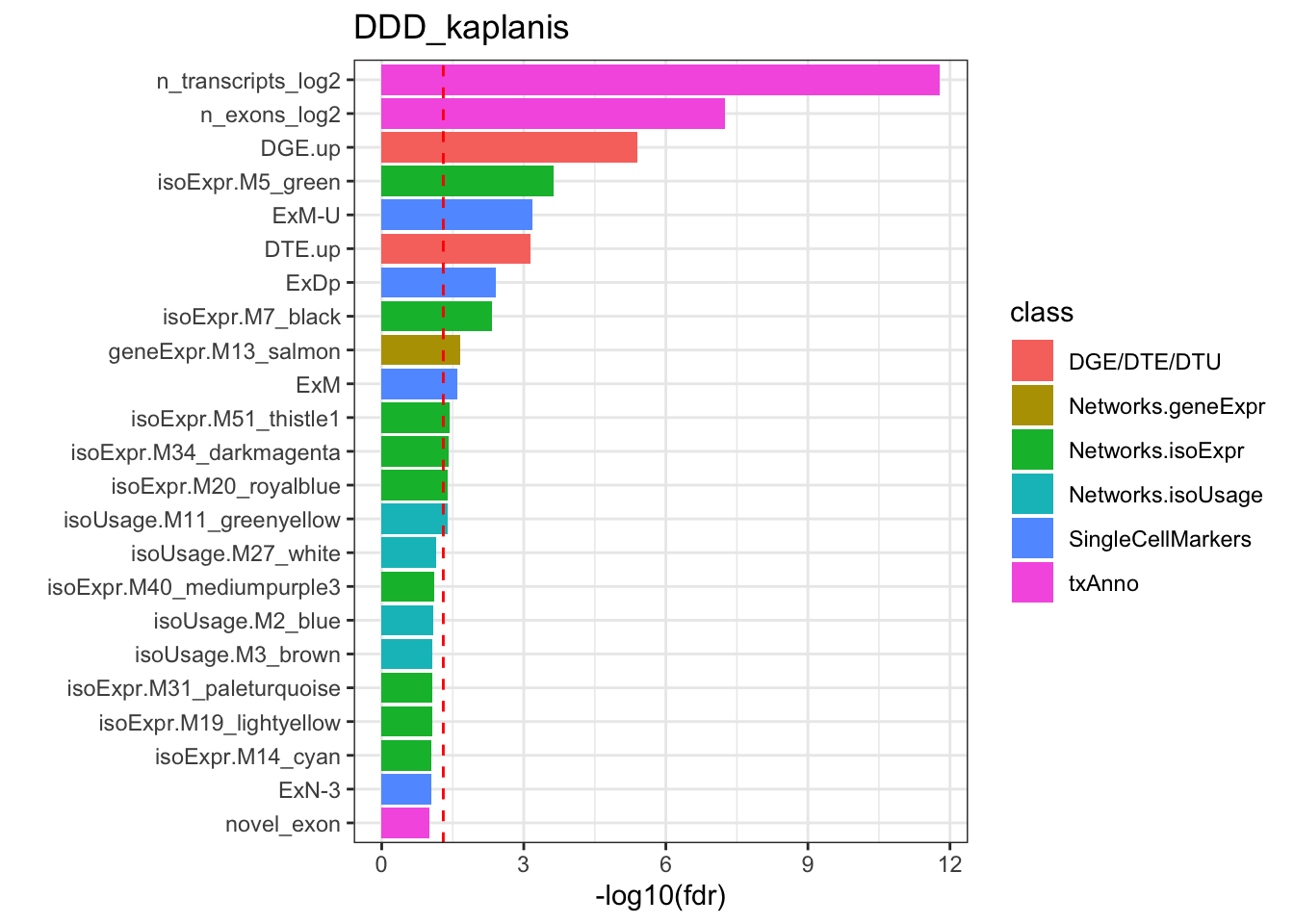
Rows: 209812 Columns: 11
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (7): network, transcript_id, module.color, module, gene_id, gene_name, ensg
dbl (3): module.number, kME, hub.rank
lgl (1): hub
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Load Rare Variant datasets
geneAnno = rtracklayer::import("ref/gencode.v33lift37.annotation.gtf.gz") %>%as_tibble() %>%filter(type=='gene')geneAnno$ensg =substr(geneAnno$gene_id,1,15)geneAnno = geneAnno[match(unique(geneAnno$ensg), geneAnno$ensg),]#Binary vector 0 for all protein coding genes, NA for non-coding; will replace with 1 for rare variant disease risk geneprotein_coding_bg =rep(NA, times=nrow(geneAnno))protein_coding_bg[geneAnno$gene_type=="protein_coding"] =0names(protein_coding_bg) = geneAnno$ensg#Start with compiled list of risk genes from SFARI, Kaplanis,risk_genes =read.csv("ref/ASD+SCZ+DDD_2022.csv")rareVar.binary =data.frame(ASD.SFARI.1 = protein_coding_bg)rownames(rareVar.binary)= geneAnno$ensgrareVar.binary$ASD.SFARI.1[geneAnno$gene_name %in% risk_genes$Gene[risk_genes$Set=="ASD (SFARI score 1)"]] =1rareVar.binary$ASD.SFARI.1or2 = protein_coding_bgrareVar.binary$ASD.SFARI.1or2[geneAnno$gene_name %in% risk_genes$Gene[risk_genes$Set=="ASD (SFARI score 1or2)"]] =1rareVar.binary$ASD.SFARI.S = protein_coding_bgrareVar.binary$ASD.SFARI.S[geneAnno$gene_name %in% risk_genes$Gene[risk_genes$Set=="ASD (SFARI syndromic)"]] =1rareVar.binary$DDD.kaplanis = protein_coding_bgrareVar.binary$DDD.kaplanis[geneAnno$gene_name %in% risk_genes$Gene[risk_genes$Set=="DDD (Kaplanis et al. 2019)"]] =1epilepsy_helbig = openxlsx::read.xlsx('http://epilepsygenetics.net/wp-content/uploads/2023/01/Channelopathist_genes_internal_2023_v2.xlsx')rareVar.binary$EPI.helbig = protein_coding_bgrareVar.binary$EPI.helbig[geneAnno$gene_name %in% epilepsy_helbig$Gene] =1# Fu et al., Nat Genetics 2022 -- ASD, NDD TADAfu=openxlsx::read.xlsx(('https://static-content.springer.com/esm/art%3A10.1038%2Fs41588-022-01104-0/MediaObjects/41588_2022_1104_MOESM3_ESM.xlsx'),'Supplementary Table 11')fu$p_TADA_ASD[fu$p_TADA_ASD==0] =min(fu$p_TADA_ASD[fu$p_TADA_ASD >0],na.rm=T)fu$p_TADA_NDD[fu$p_TADA_NDD==0] =min(fu$p_TADA_NDD[fu$p_TADA_NDD >0],na.rm=T)rareVar.logit = rareVar.binaryrareVar.logit$ASD.fuTADA=-log10(fu$p_TADA_ASD)[match(geneAnno$ensg, fu$gene_id)]rareVar.binary$ASD.fuTADA=as.numeric(fu$FDR_TADA_ASD[match(geneAnno$ensg, fu$gene_id)] < .1)rareVar.logit$NDD.fuTADA =-log10(fu$p_TADA_NDD)[match(geneAnno$ensg, fu$gene_id)]rareVar.binary$NDD.fuTADA =as.numeric(fu$FDR_TADA_NDD[match(geneAnno$ensg, fu$gene_id)] < .1)SCZ.schema =read_tsv('ref/risk_genes/SCHEMA_gene_results.tsv')
Warning: One or more parsing issues, see `problems()` for details
Rows: 18321 Columns: 26
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (8): gene_id, group, OR (PTV), OR (Class I), OR (Class II), OR (PTV) up...
dbl (16): Case PTV, Ctrl PTV, Case mis3, Ctrl mis3, Case mis2, Ctrl mis2, P ...
lgl (2): De novo mis3, De novo mis2
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 119958 Columns: 20
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (4): gene_id, group, damaging_missense_fisher_gnom_non_psych_OR, ptv_fi...
dbl (16): n_cases, n_controls, damaging_missense_case_count, damaging_missen...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 71456 Columns: 12
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (2): gene_id, group
dbl (9): xcase_lof, xctrl_lof, pval_lof, xcase_mpc, xctrl_mpc, pval_mpc, xca...
lgl (1): pval_infrIndel
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
# A tibble: 62,437 × 10
ASD.SFARI.1 ASD.SFA…¹ ASD.S…² DDD.k…³ EPI.h…⁴ ASD.f…⁵ NDD.f…⁶ SCZ.s…⁷ BIP.b…⁸
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 NA NA NA NA NA NA NA NA NA
2 NA NA NA NA NA NA NA NA NA
3 NA NA NA NA NA NA NA NA NA
4 NA NA NA NA NA NA NA NA NA
5 NA NA NA NA NA NA NA NA NA
6 NA NA NA NA NA NA NA NA NA
7 0 0 0 0 0 0.0190 0.0195 NA NA
8 NA NA NA NA NA NA NA NA NA
9 NA NA NA NA NA NA NA NA NA
10 NA NA NA NA NA NA NA NA NA
# … with 62,427 more rows, 1 more variable: EPI.epi25 <dbl>, and abbreviated
# variable names ¹ASD.SFARI.1or2, ²ASD.SFARI.S, ³DDD.kaplanis, ⁴EPI.helbig,
# ⁵ASD.fuTADA, ⁶NDD.fuTADA, ⁷SCZ.schema, ⁸BIP.bipex
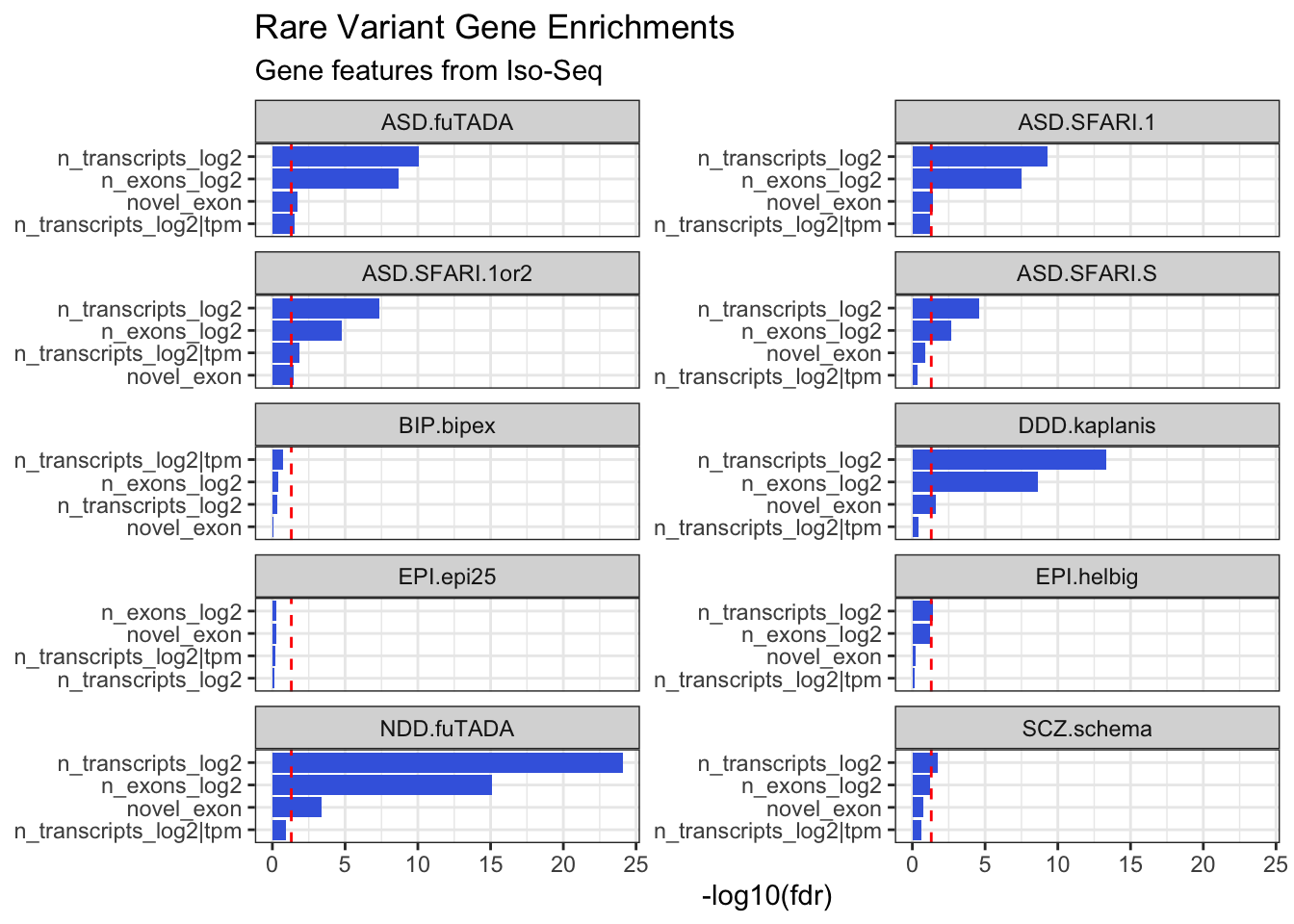
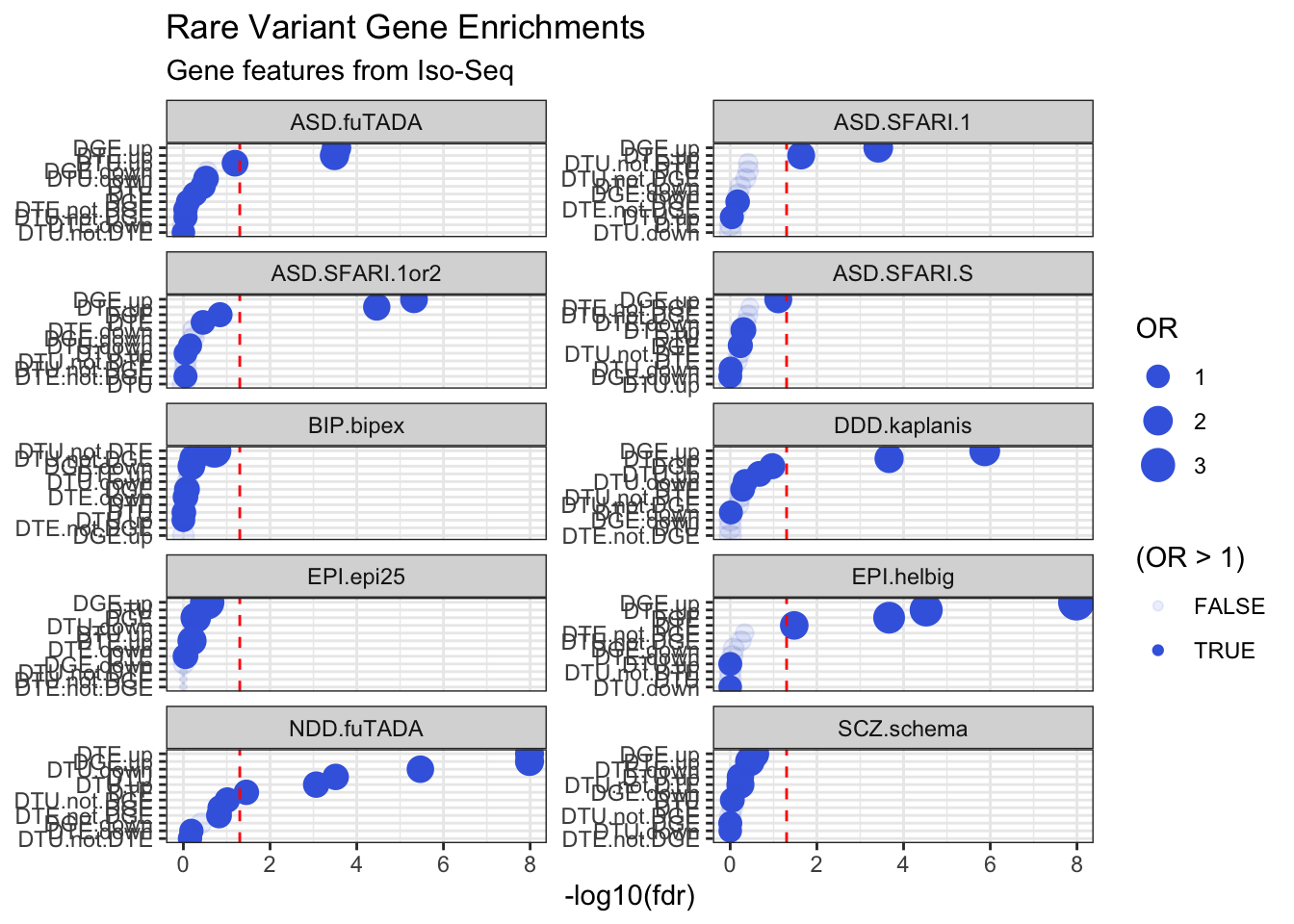
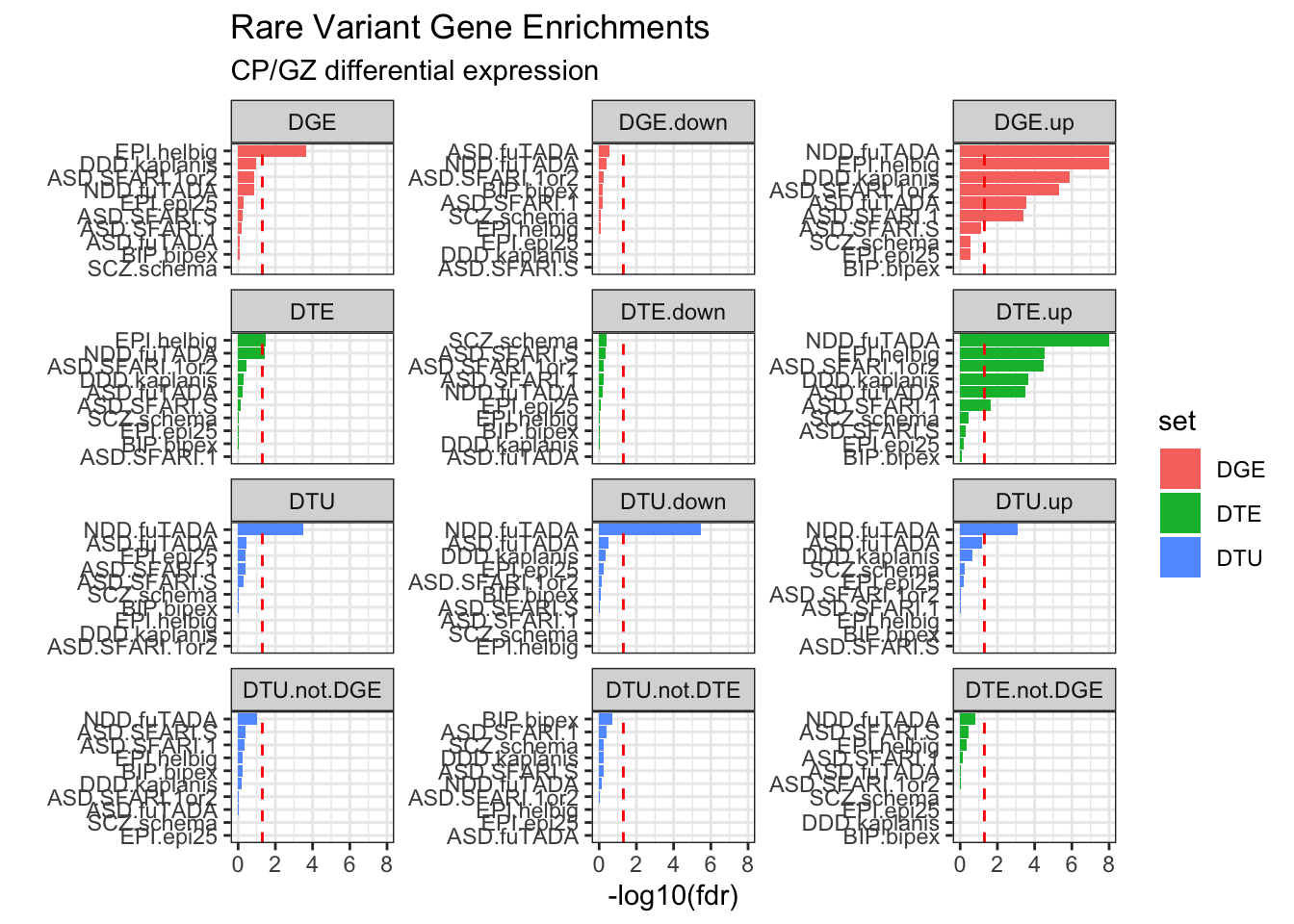
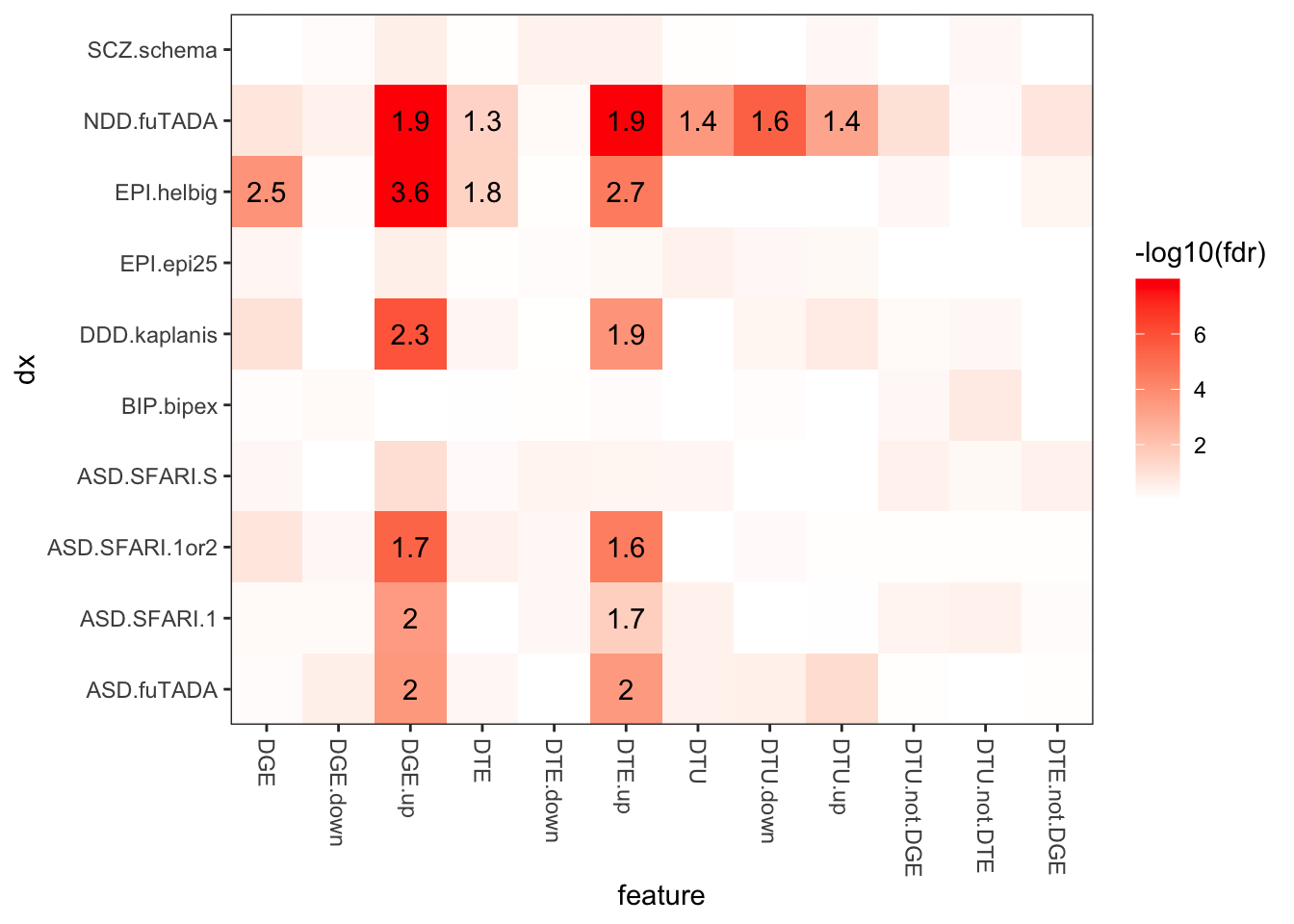
Gene Feature Annotations
# Compute fetal brain gene expressiongeneFeatures = cts %>%group_by(ensg=substr(annot_gene_id,1,15)) %>%summarise(gene_counts =sum(counts)) %>%mutate(tpm = gene_counts / (sum(gene_counts) /1000000))# Calculate number of transcripts and exons per gene in IsoSeq datageneFeatures <-as_tibble(talon_gtf) %>%mutate(ensg =str_sub(gene_id, 1, 15)) %>%group_by(ensg) %>%summarize(n_transcripts =n_distinct(na.omit(transcript_id)), n_exons =n_distinct(na.omit(exon_id))) %>%ungroup() %>%left_join(geneFeatures)
Warning: `as_data_frame()` was deprecated in tibble 2.0.0.
ℹ Please use `as_tibble()` instead.
ℹ The signature and semantics have changed, see `?as_tibble`.
Rows: 87162 Columns: 1
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): gene
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
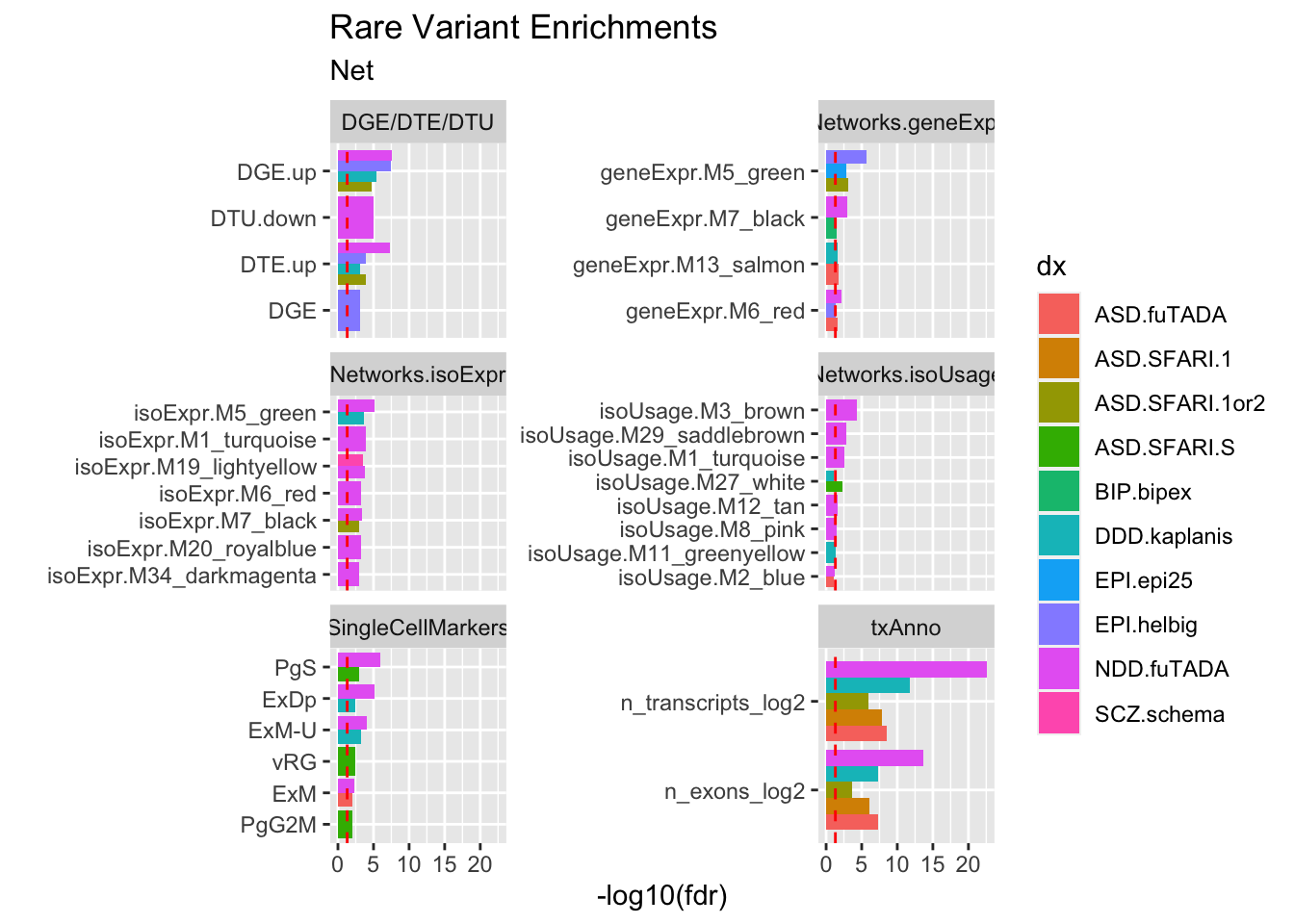
# run subsequent cells to get SingleCellDTU barif (FALSE) {ggsave("output/figures/revision1/Fig6/revisedFinal/Fig6_rareVar06.pdf", width =7, height =9)}