library(dplyr)library(ggplot2)library(ggrepel)library(gprofiler2)#Load df with averaged PDUIs across technical replicates and samples (PDUIS.averaged.stats)load("data/dapars2/PDUIS.avg.with.stats.RData")gene_names <-PDUIS.averaged.stats$Geneget_geneName <-function(tx_vec){strsplit(tx_vec, split ="|", fixed =TRUE)[[1]][2]}PDUIS.averaged.stats$geneNames <-sapply(PDUIS.averaged.stats$Gene, get_geneName)
Paired two-sample T-tests comparing CP vs VZ transcript PDUIs, with associated ggplots.
#T-test for overall averaged statistics. t.test(PDUIS.averaged.stats$CP.PDUI, PDUIS.averaged.stats$VZ.PDUI, paired =TRUE)
Paired t-test
data: PDUIS.averaged.stats$CP.PDUI and PDUIS.averaged.stats$VZ.PDUI
t = 23.028, df = 9895, p-value < 2.2e-16
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
0.01068873 0.01267778
sample estimates:
mean of the differences
0.01168326
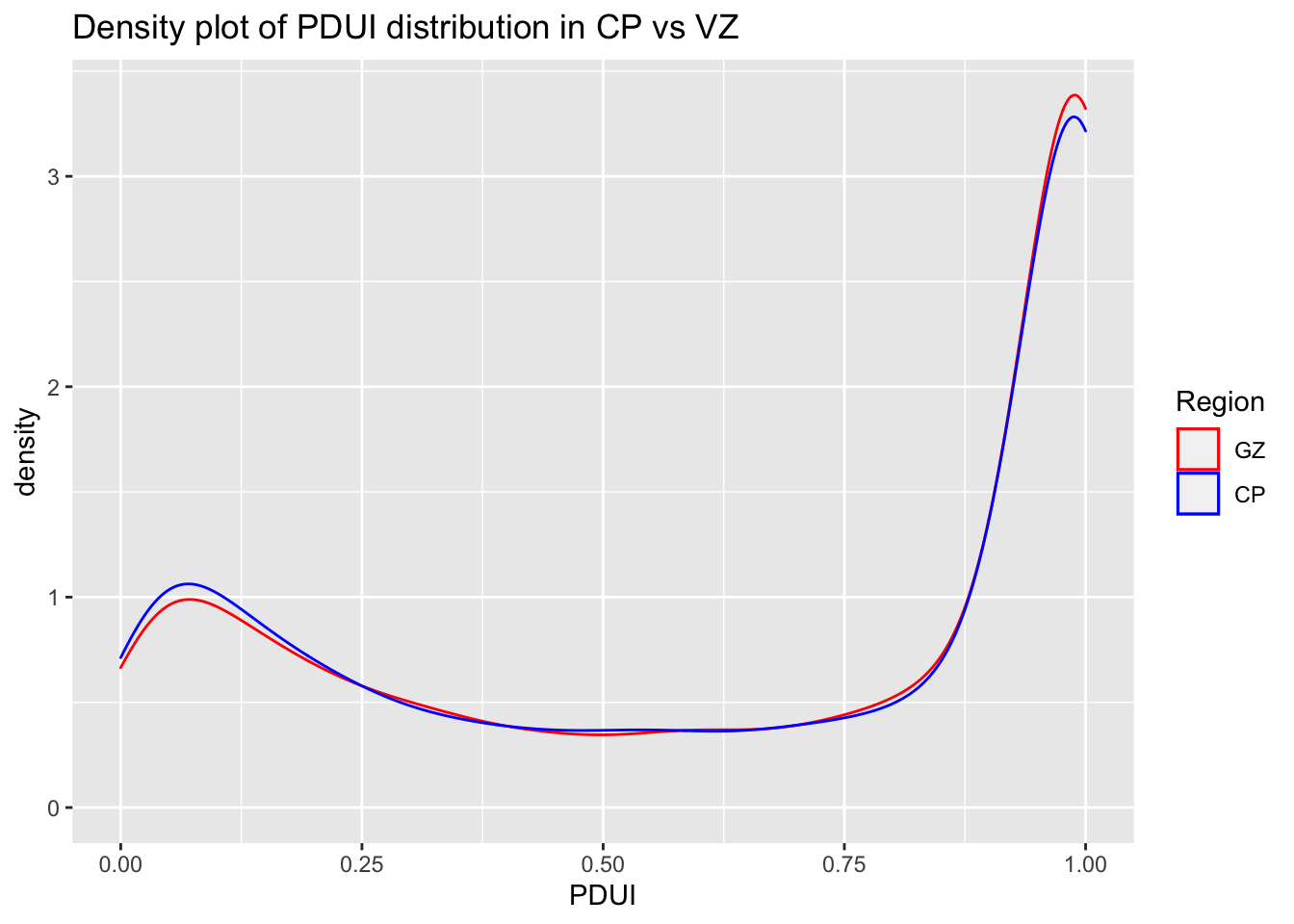
#Density plot of PDUI distribution in CP vs VZggplot(data = PDUIS.averaged.stats) +geom_density(mapping =aes(x = CP.PDUI, color ="red")) +geom_density(mapping =aes(x = VZ.PDUI, color ="blue")) +scale_color_manual(name ="Region", values =c('red'='red', 'blue'='blue'), labels =c('GZ', 'CP')) +labs(title ="Density plot of PDUI distribution in CP vs VZ", x ="PDUI")
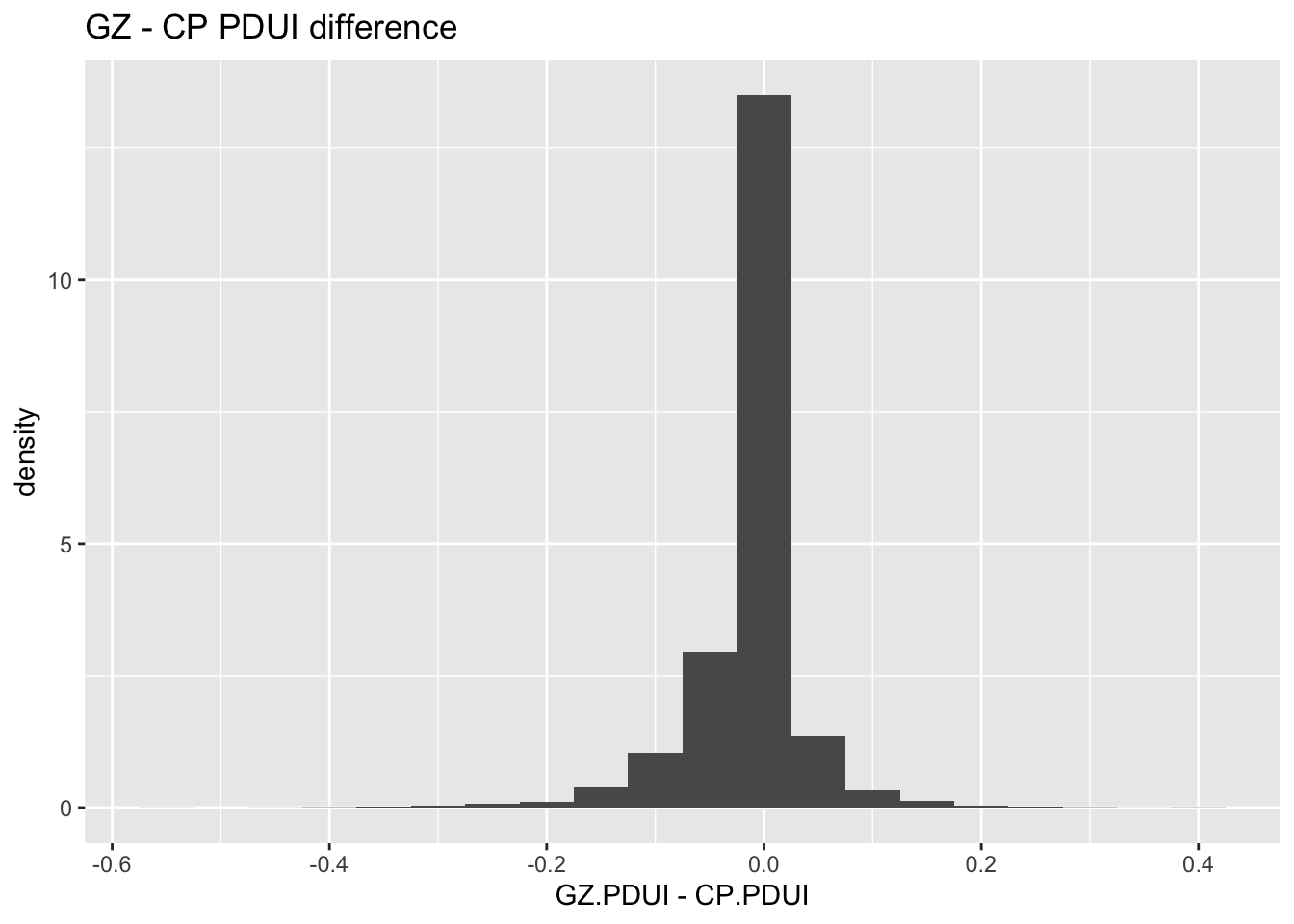
#Histogram of distributions of VZ - CP differenceggplot(data = PDUIS.averaged.stats) +geom_histogram(mapping =aes(x = vz.cp.diff, y =after_stat(density)), binwidth =0.05) +labs(title ="GZ - CP PDUI difference", x ="GZ.PDUI - CP.PDUI")
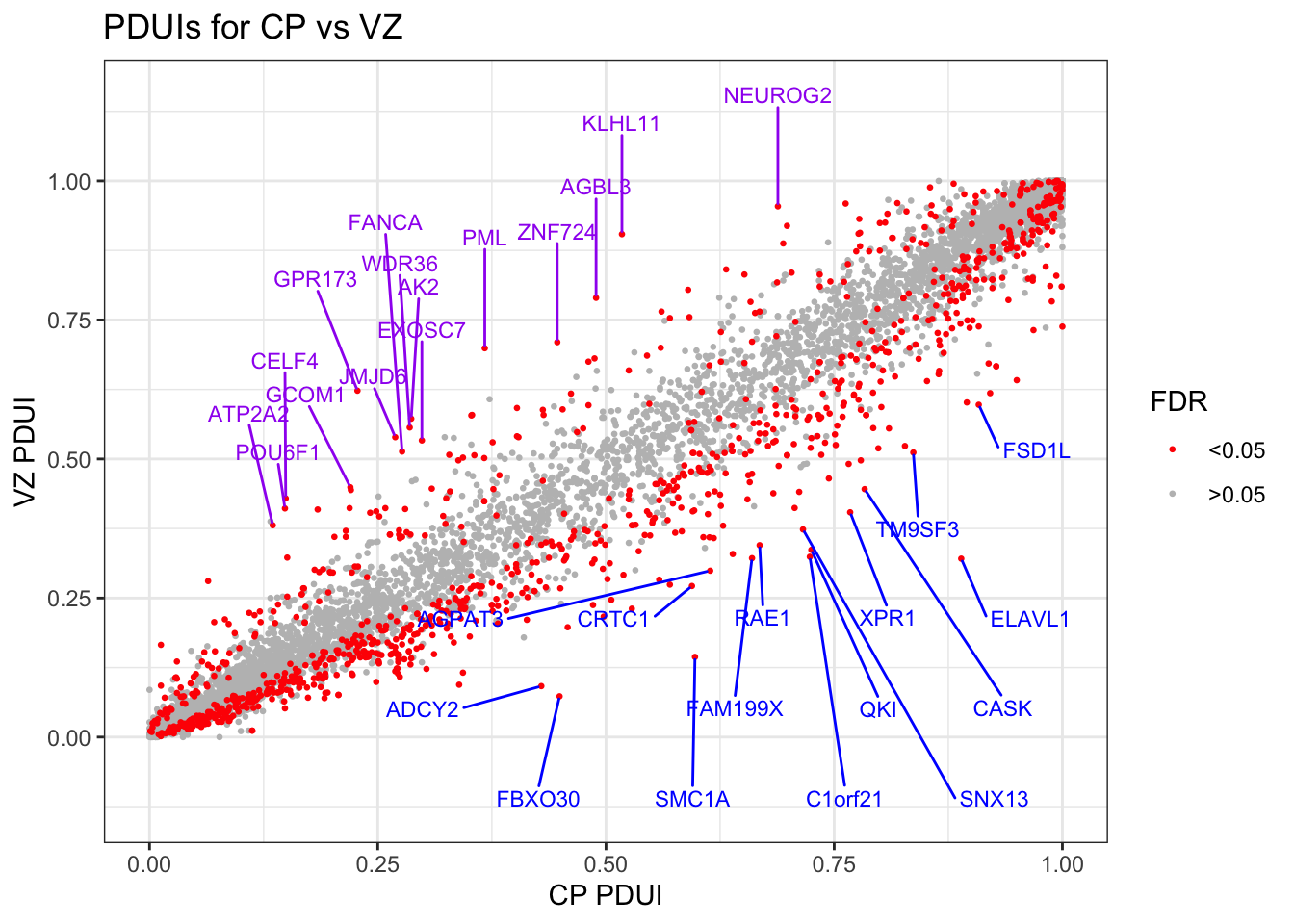
Plot CP vs VZ PDUI, denote transcripts with significant difference (FDR < 0.05 as per repeat measures ANOVA)
#Get transcripts with significantly different PDUIs based on FDR cutoff of 0.05, separate to increased or decreased PDUI in CP compared to VZ. vz.cp.neg.sig = PDUIS.averaged.stats %>%filter(FDR <0.05, vz.cp.diff <0)vz.cp.pos.sig = PDUIS.averaged.stats %>%filter(FDR <0.05, vz.cp.diff >0)vz.cp.neg.sig <-arrange(vz.cp.neg.sig, by = FDR)vz.cp.pos.sig =arrange(vz.cp.pos.sig, by = FDR)#Get top 10 genes from vz.cp.neg.sig and vz.cp.pos.sigvz.cp.pos.sig <- vz.cp.pos.sig %>%arrange(-vz.cp.diff)vz.cp.neg.sig <- vz.cp.neg.sig %>%arrange(vz.cp.diff)neg.sig.top10 <- vz.cp.neg.sig[1:15,]pos.sig.top10 <- vz.cp.pos.sig[1:15,]Fig3D=ggplot(data = PDUIS.averaged.stats) +geom_point(mapping =aes(x = CP.PDUI, y = VZ.PDUI, color =cut(FDR, c(0, 0.05, Inf))), size =0.5) +scale_color_manual(values=c("red", "gray"), labels =c("<0.05", ">0.05")) +labs(title ="PDUIs for CP vs VZ", x ="CP PDUI", y ="VZ PDUI", color ="FDR") +geom_text_repel(data = neg.sig.top10, mapping =aes(x = CP.PDUI, y = VZ.PDUI, label = geneNames), color ="blue", min.segment.length =0, box.padding =1, max.overlaps =Inf, size=3,force =20, nudge_y =-.2) +geom_text_repel(data = pos.sig.top10, mapping =aes(x = CP.PDUI, y = VZ.PDUI, label = geneNames), color ="purple", min.segment.length =0, box.padding =0, max.overlaps =Inf, size=3,force =20, nudge_y = .2) +theme_bw()Fig3D
Detected custom background input, domain scope is set to 'custom'
No results to show
Please make sure that the organism is correct or set significant = FALSE
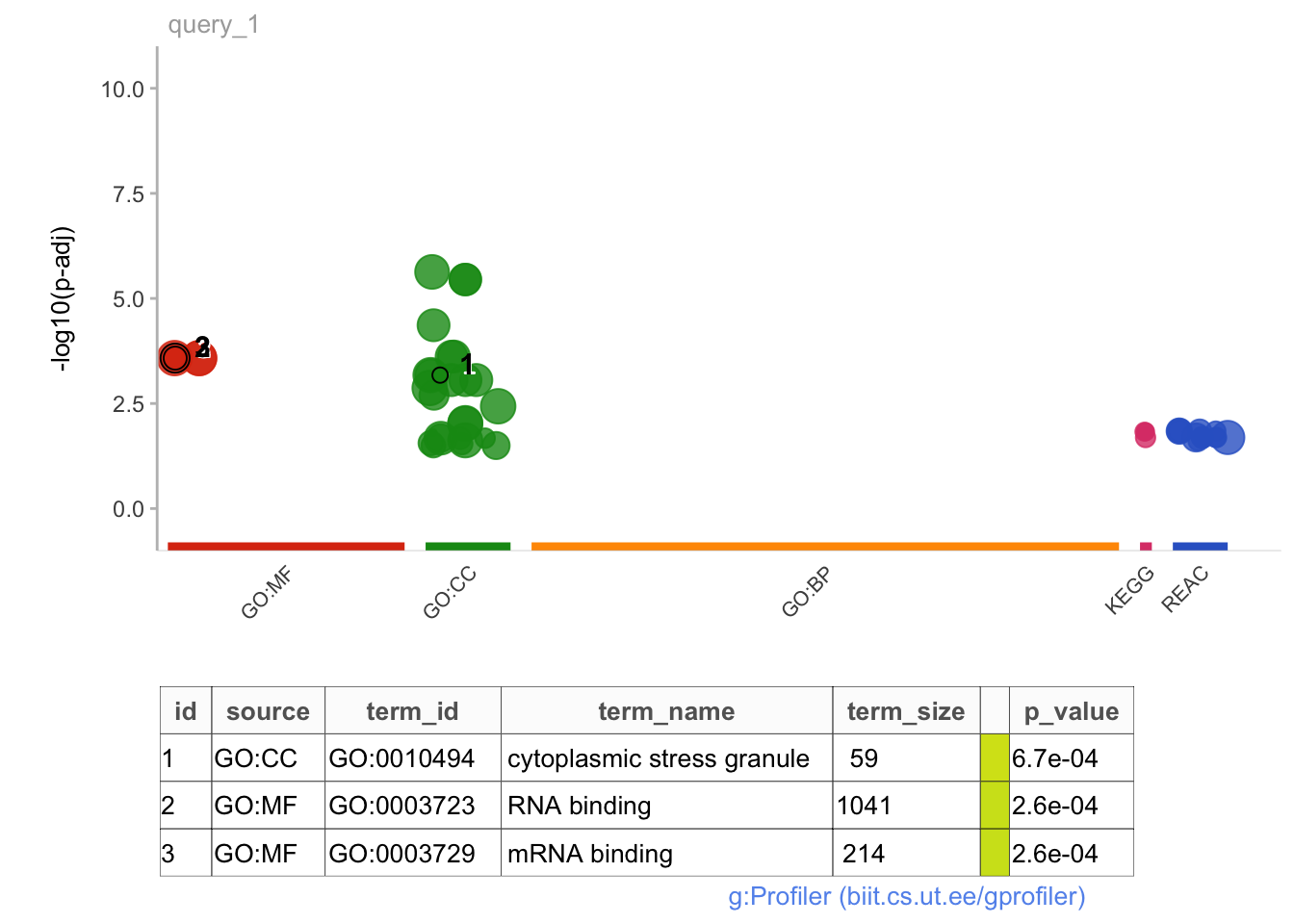
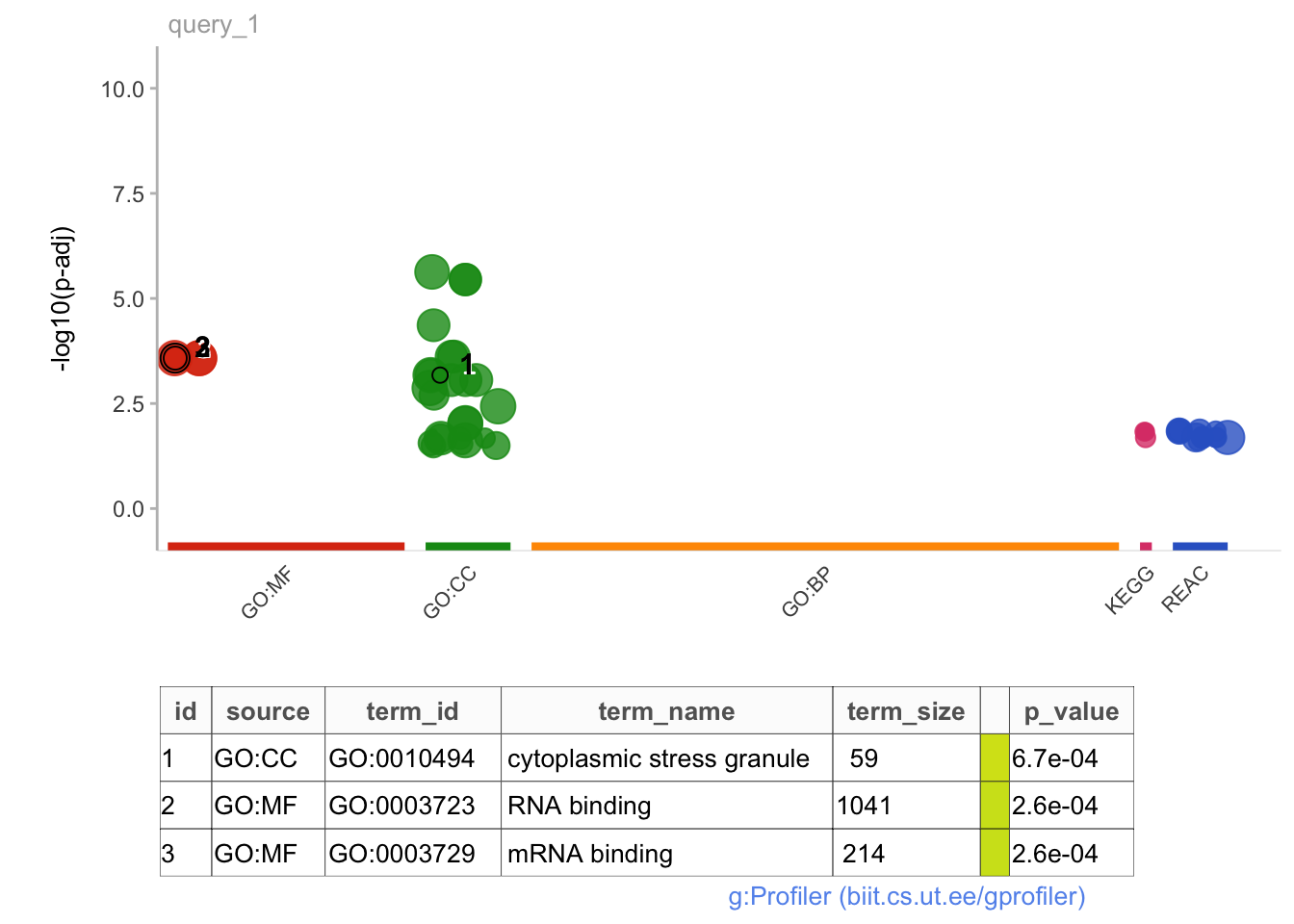
# No significant pathway enrichments from genes with significantly longer PDUI in VZ vs CPgostres_all <-gost(query = all.sig$geneNames, organism ="hsapiens", ordered_query =FALSE, significant =TRUE, custom_bg = PDUIS.averaged.stats$geneNames, correction_method ="fdr", sources =c("GO", "KEGG", "REAC"))
Detected custom background input, domain scope is set to 'custom'
# No significant pathway enrichments when genes are ranked in order of decreasing magnitude of PDUI change between brain regions#Working with all significant genes:gostplot(gostres_all, capped =FALSE, interactive = T)
Overrepresentation analysis: are transcripts w/significantly different PDUIs enriched for RBPs & TFs compared to background genes? Use RBP list from Gebauer et al. 2021
#Load RBP gene list from Gebauer et al. 2021RBPs <-scan("ref/RBPs/rbps_gebauer_nrg_2021.txt", what ="", sep ="\n")#Get background genes (transcripts with no significant change in PDUI)PDUIS.averaged.bckgrd <- PDUIS.averaged.stats %>%filter(FDR >=0.05)bckgrd.rbp <-nrow(subset(PDUIS.averaged.bckgrd, geneNames %in% RBPs))bckgrd.nonrbp <-nrow(subset(PDUIS.averaged.bckgrd, !geneNames %in% RBPs))sig.rbp <-nrow(subset(PDUIS.averaged.stats %>%filter(FDR <0.05), geneNames %in% RBPs))sig.nonrbp <-nrow(subset(PDUIS.averaged.stats %>%filter(FDR <0.05), !geneNames %in% RBPs))#Run one-sided Fisher's exact testd <-data.frame(bckgrd =c(bckgrd.rbp, bckgrd.nonrbp), sig =c(sig.rbp, sig.nonrbp))row.names(d) <-c("RBP", "non-RBP")d
bckgrd sig
RBP 3074 453
non-RBP 5809 560
fisher.test(d, alternative ="less")
Fisher's Exact Test for Count Data
data: d
p-value = 2.06e-10
alternative hypothesis: true odds ratio is less than 1
95 percent confidence interval:
0.0000000 0.7321337
sample estimates:
odds ratio
0.6541755