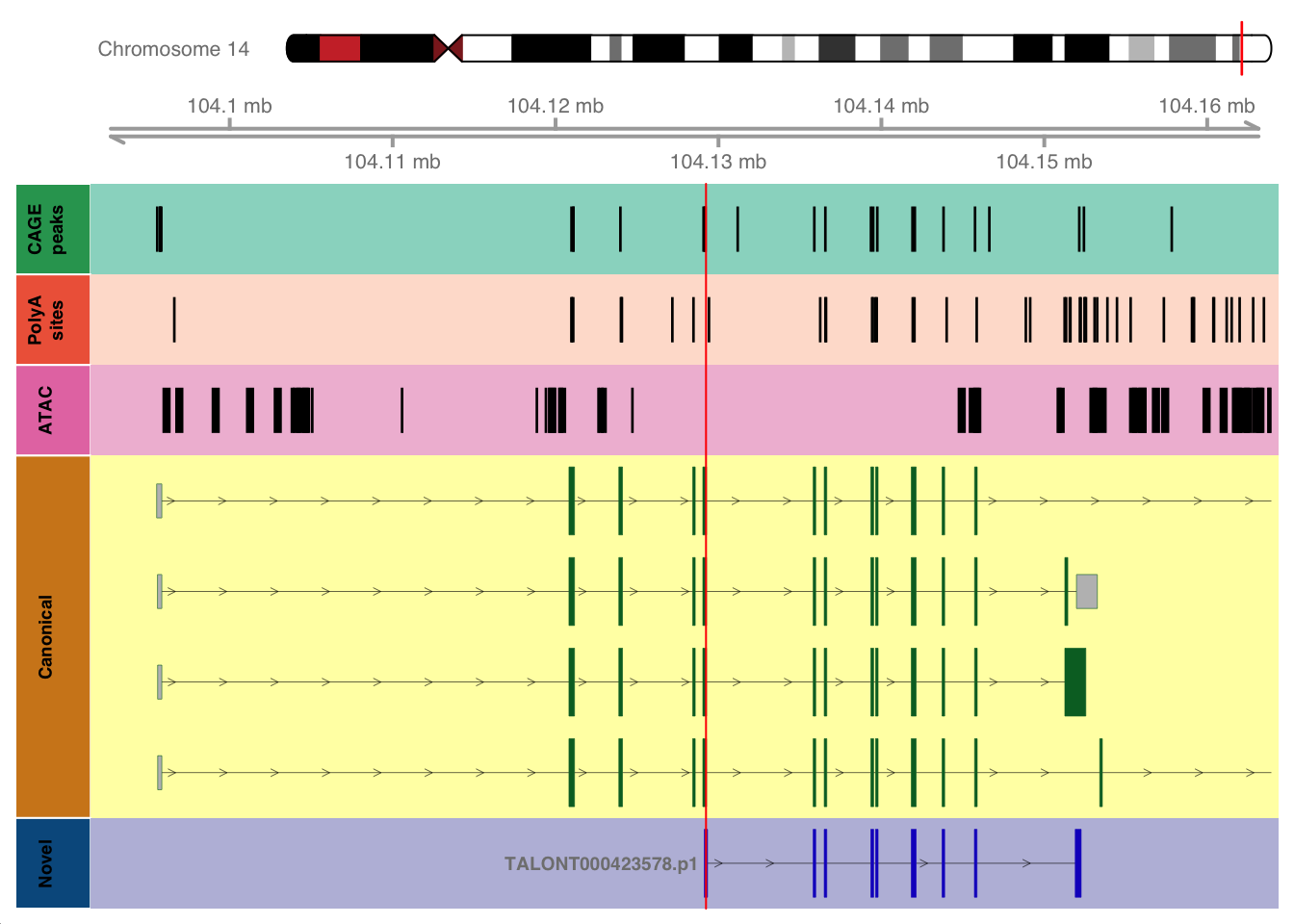
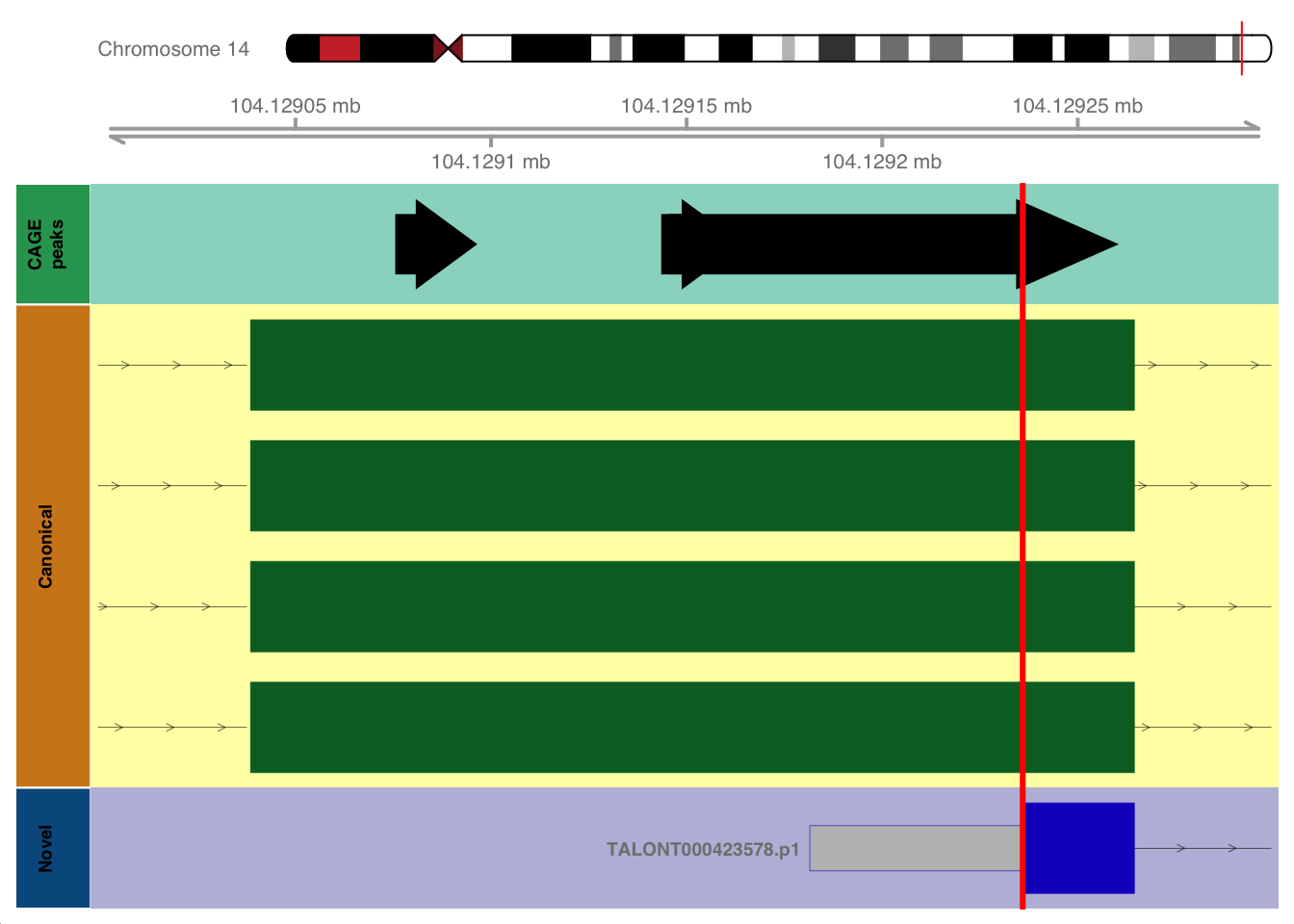
The following objects are masked from 'package:base':
expand.grid, I, unname
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: grid
library(GenomicFeatures)
Loading required package: AnnotationDbi
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Import genomic features from the file as a GRanges object ... OK
Prepare the 'metadata' data frame ... OK
Make the TxDb object ...
Warning in .get_cds_IDX(mcols0$type, mcols0$phase): The "phase" metadata column contains non-NA values for features of type
stop_codon. This information was ignored.
Warning in .reject_transcripts(bad_tx, because): The following transcripts were dropped because they have incompatible
CDS and stop codons: ENST00000422803.2_2, ENST00000618549.1_2,
ENST00000619291.4_2, ENST00000621077.1_2, ENST00000621229.1_2,
ENST00000631326.2_2
known_transcript=GRanges()for (i in known){if (!i %in%names(gencode_cds)){cat("Known transcript is not coding:",i)next } thistranscript=unlist(gencode_cds[i,])elementMetadata(thistranscript)=NULLelementMetadata(thistranscript)$feature="cds" known_transcript=c(known_transcript,thistranscript)if (i %in%names(gencode_5utr)){ thistranscript=unlist(gencode_5utr[i,])elementMetadata(thistranscript)=NULLelementMetadata(thistranscript)$feature="utr" known_transcript=c(known_transcript,thistranscript) }if (i %in%names(gencode_3utr)){ thistranscript=unlist(gencode_3utr[i,])elementMetadata(thistranscript)=NULLelementMetadata(thistranscript)$feature="utr" known_transcript=c(known_transcript,thistranscript) }}novel_transcript=GRanges()for (i in novel){if (!i %in%names(isoseq_cds)){cat("Novel transcript is not coding:",i)next } thistranscript=unlist(isoseq_cds[i,])elementMetadata(thistranscript)=NULLelementMetadata(thistranscript)$feature="cds" novel_transcript=c(novel_transcript,thistranscript)if (i %in%names(isoseq_5utr)){ thistranscript=unlist(isoseq_5utr[i,])elementMetadata(thistranscript)=NULLelementMetadata(thistranscript)$feature="utr" novel_transcript=c(novel_transcript,thistranscript) }if (i %in%names(isoseq_3utr)){ thistranscript=unlist(isoseq_3utr[i,])elementMetadata(thistranscript)=NULLelementMetadata(thistranscript)$feature="utr" novel_transcript=c(novel_transcript,thistranscript) }}known_chr=as.character(known_transcript@seqnames)[1]novel_chr=as.character(novel_transcript@seqnames)[1]if (known_chr == novel_chr){ chr=known_chr}else{stop("chr differs between known and novel")}known_strd=as.character(known_transcript@strand)[1]novel_strd=as.character(novel_transcript@strand)[1]if (known_strd == novel_strd){ strd=known_strd}else{stop("strand differs between known and novel")}